Article Text

Abstract
Cerebral neurovascular development is a complex and coordinated process driven by the changing spatial and temporal metabolic demands of the developing brain. Familiarity with the process is helpful in understanding neurovascular anatomic variants and congenital arteriovenous shunting lesions encountered in endovascular neuroradiological practice. Herein, the processes of vasculogenesis and angiogenesis are reviewed, followed by examination of the morphogenesis of the cerebral arterial and venous systems. Common arterial anatomic variants are reviewed with an emphasis on their development. Finally, endothelial genetic mutations affecting angiogenesis are examined to consider their probable role in the development of three types of congenital brain arteriovenous fistulas: vein of Galen malformations, pial arteriovenous fistulas, and dural sinus malformations.
- arteriovenous malformation
- artery
- congenital
- genetic
- vein
Statistics from Altmetric.com
Development of the cerebrovascular system is a complex yet reproducible process with numerous spatially and temporally coordinated events. Early anatomic descriptions made by Streeter1 and Padget2 3 provide the foundational knowledge used today, with further insights from radiological correlates advanced by Lasjaunias4 5 and Raybaud.6–8 Cerebral neurovascular development can be divided into two parts: vasculogenesis followed by angiogenesis. Herein, a timeline of events of typical development is reviewed and examined in order to explain the presentation of anatomic variants and congenital intracranial arteriovenous shunting lesions that are encountered in endovascular neuroradiological practice.
Vasculogenesis
The first endothelial tubes formed by a single layer of endothelium are constituted during vasculogenesis, which begins with the differentiation of multipotent mesodermal cells into endothelial cell (EC) precursor angioblasts.9 Some of these angioblasts migrate to the extra-embryonic yolk sac and cluster with hematopoietic stem cells to form hemangioblastic aggregates. These organize into blood islands, which are collections of hematopoietic stem cells surrounded by ECs, that coalesce to form a primitive vascular network.10 Other angioblasts remain intra-embryonic and organize into a long axial precursor vessel that undergoes arterial/venous segregation to form the paired dorsal aorta and cardinal vein.11 12 Connections exist between the primitive longitudinal vessels and the primordial capillary plexus, which extends intra-embryonally and cranially to invade the solid mesenchyme of the meninx primitiva surrounding the closed cranial neural pore at 4 weeks post-conception. Shortly after, endothelial channels proliferate to form the perineural vascular network of the meninx primitiva, which has superficial and deep layers. The superficial (dural) plexiform vascular network communicates with the paired dorsal aorta and cardinal veins, while the deep layer (pial) resembles a capillary network. Communicating segments exist between the layers (arachnoid) that later organize into the recognizable pattern of surface brain arteries and veins. The meninx primitiva overlying the forebrain (anterior neural plate) is of neural crest origin, giving rise to later anterior circulation autonomic control, while the remainder is from somitic mesoderm.8 The EC differentiation of angioblasts is regulated by tissue-derived factors, including vascular endothelial growth factor (VEGF), fibroblast growth factor 2, and bone morphogenic protein 4 (BMP4).9
Initially extrinsic diffusion of nutrients from the periphery can supply the neural tube to meet its metabolic requirements. At this point the primordial vascular plexus in the meninx primitiva contains vessels that remain plastic with respect to arterial or venous specification, although genetic factors define an arterial or venous identity for some ECs even at the angioblast stage before the first heart beat.12 This specification has been shown to be reversible epigenetically via hemodynamic stimuli that become more pronounced throughout development.8 12 Differential expression of several molecular markers identify vessel fate as arterial or venous prior to assembly of the vessel wall. Arterial specific markers include signaling molecules ephrin-B2, neuropilin-1, Notch3, DLL4, and gridlock, while venous markers include EphB4 (the receptor for ephrin-B2) and chicken ovalbumin upstream promoter-transcription factor II, the latter thought to downregulate pro-arterial Notch signaling.10 12 13
Angiogenesis
As the thickness of the neural tube increases to >100–200 microns and the metabolic activity of the developing neural tissue increases, extrinsic diffusion of nutrients and oxygen from the primitive vasculature of the meninx primitiva becomes insufficient.8 10 This provides stimulus for the formation of primordial choroid plexuses between weeks 5 and 7, whereby portions of the primitive vasculature involute into the neural canal at telencephalic and rhombencephalic locations.14 15 The development of the choroid plexuses enables further diffusive delivery of nutrients to neural tissue in periventricular locations via cerebrospinal fluid transport. However, metabolic demands will rise further owing to increases in cellular proliferation in the germinal matrices, followed by cortical migration and organization that expands the fetal brain, with an emerging need for an intrinsic vascular system.8
Angiogenesis is the growth of new vessels from existing ones following a complex and regulated process.16 There are two mechanisms by which angiogenic vessels arise: sprouting and intussusception. Most research to date has involved extravascular sprouting angiogenesis, and many details regarding angiogenic intussusception—the intravascular ‘splitting’ of existing vessels through internal division that can be used for vessel pruning, modifying bifurcation angulation, or new vessel formation—remain unknown.17 As metabolic demands increase within the developing germinal centers, hypoxia-induced growth factors (the most significant being VEGFA) are released from the periventricular zones with their gradients stimulating the sprouting of cords of ECs from the superficial vascular plexus into the ischemic tissue.8 18 This elaborate spatiotemporally coordinated process involves phenotypic changes in ECs from a quiescent phalanx cell phenotype into migrating tip cells (TCs) that bud into adjacent tissue and lead a trailing cord of stalk cells (SCs) that are ultimately responsible for vessel maturation and formation of a lumen.16 19 Endothelial cords extend from the surface vasculature to form patent transcerebral loops near the deep locations of ischemic tissue and provide hemodynamic relief of regional hypoxia. As the cerebral hemispheres expand during development, the circumference of the cortex increases at a rate greater than the periventricular surface, giving the vasculature its characteristic wedge-shaped pattern.20
Morphogenesis of the ascending aortic arch and great vessels
The heart initially forms in a cephalic location and as it descends into the thorax, its connection through the ventral aortic sac with the paired dorsal aorta undergoes multiple changes that produce the adult configuration of the aortic arch. This occurs through the production and remodeling of six pairs of branchial arches, necessary to navigate the developing pharyngeal pouches.2 21
The first branchial arch artery forms at a crown–rump length of ~1.3 mm. It regresses by 4 mm, with the remnant forming the mandibular artery that accompanies the superior petrosal nerve and eventually becomes the vidian artery.22
The second branchial arch artery forms between 3 mm and 4 mm and makes ventral and dorsal divisions, the latter connecting to the cranial dorsal aorta that becomes the internal carotid artery (ICA). The ventral portion becomes the ventral pharyngeal artery that is the precursor to the external carotid artery (ECA). The dorsal branch connected to the ICA is called the hyoid artery, which transiently develops the stapedial artery as a branch. At the end of development, the remnant of the second branchial arch artery is the caroticotympanic artery.22 23
The third and fourth branchial arch arteries appear at 4 mm. The third fuses with the distal paired dorsal aorta to form the proximal ICAs. The dorsal aorta then regresses between the third and fourth branchial arch arteries bilaterally. The fourth branchial arch artery contributes to the aortic arch on the left and proximal subclavian artery on the right.
The fifth branchial artery is one of contention, probably related to the fact that if it exists, it is only briefly, and its contribution is minimal.22
The final sixth pair of branchial arch arteries are visible between 5 mm and 7 mm, with the proximal portions contributing to their ipsilateral pulmonary arteries, while the distal portions involute at 28 mm. This signals the beginning of the post-branchial phase according to Congdon,21 which includes left dorsal aorta regression between the third and fourth arches and derivation of the subclavian arteries from the seventh intersegmental arteries of the dorsal aorta bilaterally as the final adult form of the aortic arch and great vessels manifest (figure 1 and table 1).22 23
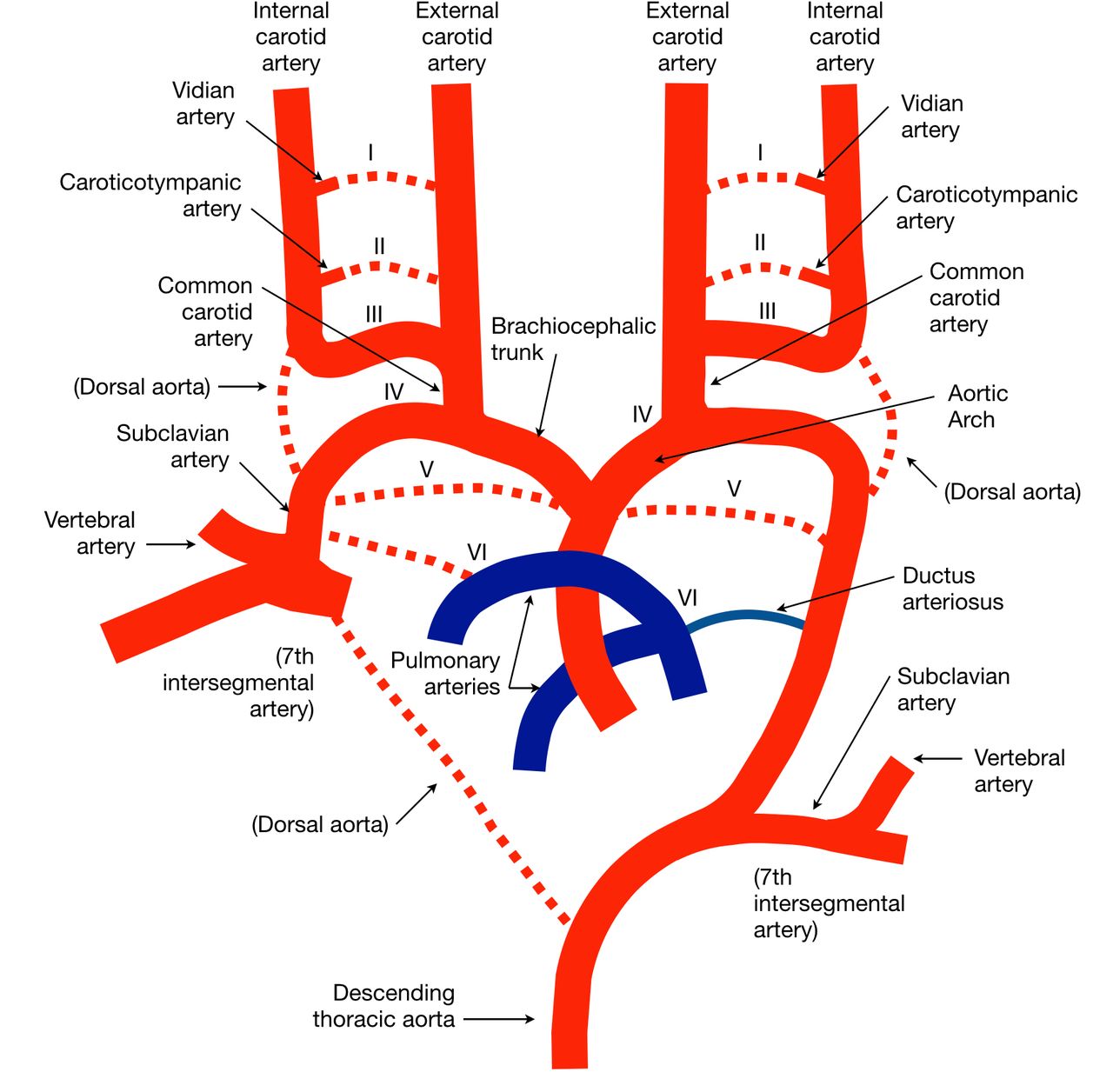
Branchial arch arteries and the final adult form of the aortic arch and origins of the great vessels. Transient vessels are represented with dashed lines. see text and table 1 for more details.
Remnants of the branchial arch arteries
Anatomic variations: head and neck vessels
Common origin of the brachiocephalic and left common carotid arteries
With a prevalence estimated to approach 25%, this anatomic variant results from the proximal third branchial arch artery being absorbed into the right rather than left aortic sac.23
Aberrant right subclavian artery
This variant has a prevalence of approximately 1–2%, resulting from excessive proximal right dorsal aorta regression beyond the fourth branchial arch artery to the origin of the seventh intersegmental artery.23 Sometimes termed arteria lusoria, compressive symptoms on adjacent structures, most notably the esophagus and trachea, can require surgical intervention in some cases.
Carotid aplasia or hypoplasia
As listed in table 2, the ICA can be divided into seven segments based on their embryologic origins as described by Lasjaunias and Santoyo-Vazquez, which completes formation by 4 weeks.4 If there is erroneous development in any one of these segments, hypoplasia or less likely aplasia may result, with a reported incidence of 0.01% in the general population.24 Hypoplasia is commonly seen starting near the carotid bifurcation and can be inferred by a diminutive bony carotid canal as it forms at 5 weeks.22 The bony canal may be absent with carotid aplasia.
Embryologic segments of the internal carotid artery
Aberrant internal carotid artery and persistent stapedial artery
Normally the intracranial ICA originates at the entrance to the carotid canal; however, secondary to segmental cervical ICA aplasia, the inferior tympanic artery can persist from the ascending pharyngeal artery and hypertrophy to deliver blood through the caroticotympanic artery to the carotid siphon. With a characteristic retrotympanic course through the middle ear, the differential includes a glomus tympanicum tumor and can lead to iatrogenic hemorrhage if not properly identified. It is more common in women and on the right side.22
The transient stapedial artery from the hyoid artery has superior and inferior divisions that later form the middle meningeal artery (MMA) and inferior alveolar artery, respectively. The inferior division normally exits the cranium through the foramen spinosum to anastomose with the ventral pharyngeal artery around 10 weeks, initiating regression of the stapedial artery, so the MMA is supplied by the ECA in adult life.23 If this process does not occur, a persistent stapedial artery (PSA) will form that supplies the MMA. Although absence of the foramen spinosum is a sign for a PSA, prevalence of an absent foramen spinosum is 3% while the PSA has a prevalence of 0.5%.23 25 A PSA can occur in isolation or with an aberrant carotid artery.
Morphogenesis of surface brain arteries
Following vasculogenesis, the primitive vascular plexus undergoes remodeling, leading to a pattern of surface vessels in response to spatiotemporal cues from the changing metabolic demands of the developing brain. Padget divided the patterning of the superficial cranial vessels into seven stages that are briefly reviewed here and illustrated in figure 2.2
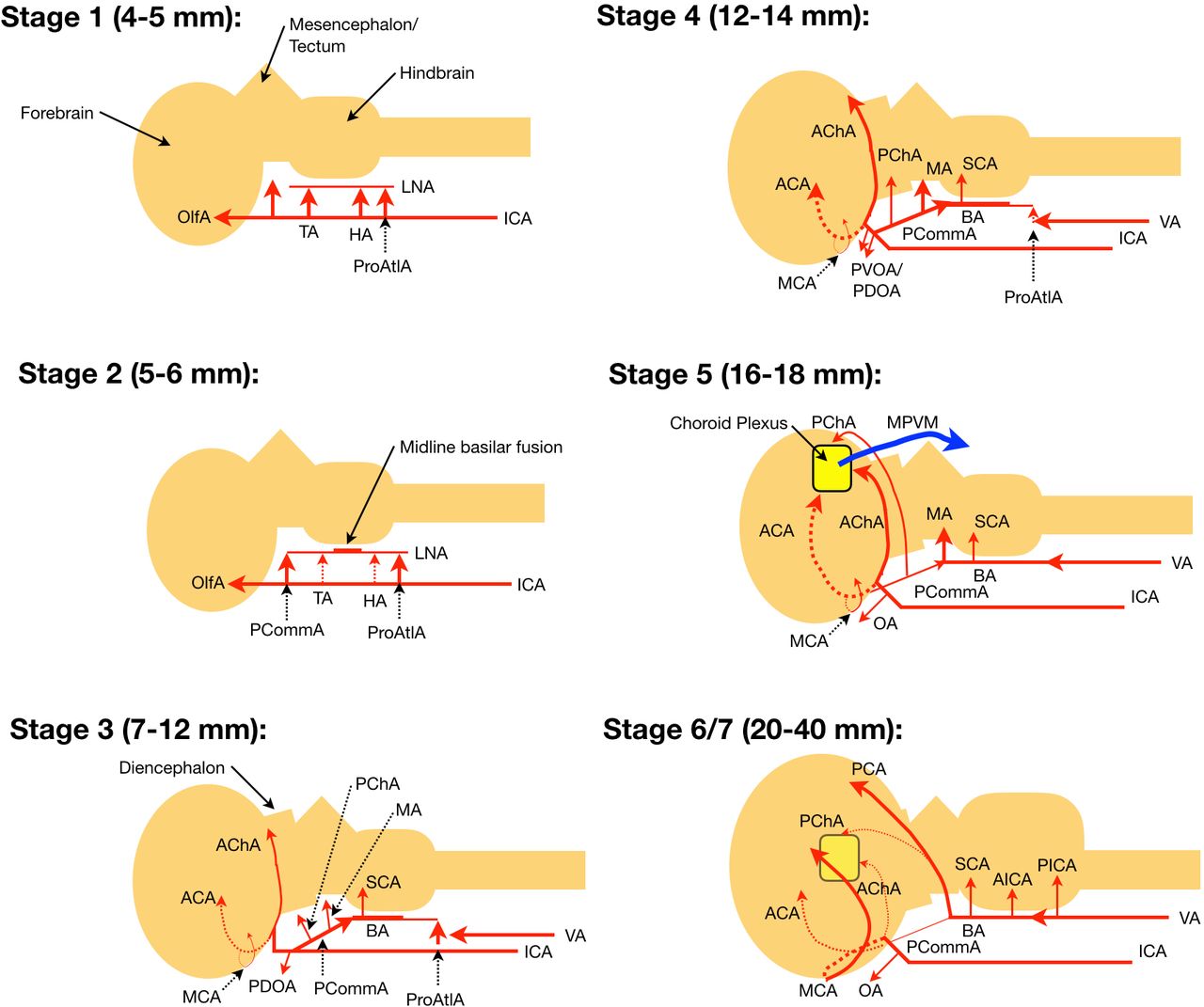
Stages of arterial morphogenesis according to Padget. Depictions are from a sagittal view, with vessels behind the cerebral hemisphere (in the midline) represented by dashes. Sizes provided refer to crown–rump length. Stage 5 corresponds to the ‘choroid stage’ where arteriovenous fistulization occurs in vein of Galen malformations between the choroidal and mesencephalic arterial feeders and median prosencephalic vein of Markowski. See the text for details. ACA, anterior cerebral artery; AChA, anterior choroidal artery; AICA, anterior inferior cerebellar artery; BA, basilar artery; HA, hypoglossal artery; ICA, internal carotid artery; LNA, longitudinal neural artery; MA, mesencephalic artery; MCA, middle cerebral artery; OA, ophthalmic artery; OlfA, olfactory artery; PCA, posterior cerebral artery; PChA, posterior choroidal artery; PCommA, posterior communicating artery; PDOA, primitive dorsal ophthalmic artery; PICA, posterior inferior cerebellar artery; ProAtlA, proatlantal artery; PVOA, primitive ventral ophthalmic artery; SCA, superior cerebellar artery; TA, trigeminal artery; VA, vertebral artery.
Stage 1 (4–5 mm, 28–29 days)
The distal primitive ICA bifurcates into cranial and caudal branches. The cranial branch is the olfactory artery, which supplies the forebrain and later becomes the anterior cerebral artery (ACA), while the caudal branch supplies a plexus over the mesencephalon and later becomes the posterior communicating artery (PCommA). A connection between bilateral ICAs posterior to Rathke’s pouch forms an early anastomosis of the circle of Willis.8 The hindbrain is supplied via bilateral ventral plexular longitudinal neural arteries (LNAs) that exhibit transient cartoid-vertebrobasilar anastomoses. The trigeminal artery (TA), at the level of the trigeminal ganglion, is the most cranial anastomosis formed by presegmental arteries, followed by the hypoglossal artery (HA) at the level of the hypoglossal nerve. The caudal anastomosis, the proatlantal artery (ProAtlA), forms from the first cervical intersegmental artery and courses along the first cervical nerve.
Stage 2 (5–6 mm, 29 days)
The caudal branch of the ICA extends posteriorly to fuse with the LNAs, forming the PCommAs. The basilar artery starts to form as midline fusion of the LNAs begins. Common origins for basilar perforators are extremely rare given this fusion process.23
Stage 3 (7–12 mm, 32 days)
Forming along the base and between the developing cerebral hemispheres, the ACAs start to take shape from coalescing vascular plexus. Similarly, more posteriorly along the base of the hemispheres the primitive anterior choroidal artery (AChA) prominently takes form, with branches directed towards the diencephalon. The groups of anterior perforators (recurrent artery of Huebner (RAH), medial, and lateral lenticulostriates) form projecting laterally off the ACA, with a couple of striates coalescing over the telencephalon to form the primitive middle cerebral artery (MCA).23 More proximally, the primitive dorsal ophthalmic artery (PDOA) appears. As the extent of basilar fusion increases, two dorsal branches arise from the PCommA. Proximally the posterior choroidal artery (PChA) is directed toward the diencephalon, while distally the mesencephalic artery (MA) is directed toward the midbrain. The vertebral arteries (VAs) begin to form through fusion of the longitudinal anastomoses of the cervical intersegmental arteries at the C1 to C7 levels, with regression of the vestigial carotid-vertebrobasilar anastomoses now complete. Over the rhombencephalon the superior cerebellar artery (SCA) begins to appear, whose supply will later include the deep cerebellar nuclei.8 23
Stage 4 (12–14 mm, 35 days)
Medial branches from the ICAs form a plexus that eventually becomes the anterior communicating artery (ACommA), which in 70% forms above the optic chiasm, while the remainder are located above the cisternal segment of the optic nerves (ONs).23 The primitive dorsal and ventral ophthalmic arteries (PDOA and PVOA, respectively) are now present. The PDOA and PVOA later anastomose in the orbit near the ON, with a new anastomosis forming the ophthalmic artery origin of the ICA.26 The PDOA goes on to commonly form the inferolateral trunk while the PVOA becomes the A1 segment of the ACA. Only 20% of the time will co-dominant A1 branches form, with a dominant A1 branch supplying the two A2 branches being a more common anatomic arrangement.23 Stage 4 sees increasing prominence of the AChA, PChA, and MA as the basilar and vertebral arteries develop further.
Stage 5 (16–18 mm, 40 days)
The choroid plexuses begin to form, supplied anteriorly by the ACA, inferiorly by the AChA, and posteriorly by the PChA, defining the ‘choroid stage’ of Klosovskii. The MA forms a plexus over the midbrain and as noted by Raybaud, these four branches form the arterial feeders (the MA providing transmesencephalic feeders) for vein of Galen malformations (VoGMs).7
Stage 6 (20–24 mm, 44 days) and stage 7 (40 mm, 52 days)
The MCA and lenticulostriate arteries complete formation, including the RAH, arising laterally from the ACA. The dorsal branch of the MA annexes the classic vascular territory of the posterior cerebral artery (PCA) from the AChA, converting the supply of the inferior temporal and medial occipital regions from the carotid to vertebrobasilar system. In the posterior fossa the anterior inferior cerebellar artery (AICA) and posterior inferior cerebellar artery (PICA) are now identified.8
Anatomic variations: surface brain arteries
Fenestrations
Incomplete fusion during cerebrovascular development producing a fenestration has a reported frequency with CT angiography of between 10% and 15%27 28 and most commonly affects the ACommA (40% at autopsy, ~5–7% on CT angiography)23 27 28 and basilar artery (5% at autopsy,~2–6% on CT angiography).27–29 MCA fenestrations (0.5–1% prevalence) were noted in five cases by Gailloud et al to coexist with an inferior temporopolar branch, suggesting failed early perforator regression.30 Fenestrations have been suggested to more commonly involve the posterior circulation,31 with basilar fenestrations believed to predispose aneurysm formation due to altered flow dynamics and the local defect in the media of the vessel wall.32 33
Persistent vestigial arteries
The most common persistent vestigial artery (PVA) is the persistent TA (85% of all PVAs, and seen on 0.1–0.6% of angiographic scans, with a 12–16% prevalence of PHACE syndrome), followed by the persistent HA (seen on 0.02–0.1% of angiographic scans).23 The sella turcica is traversed by ~50% of persistent TAs, which forms a connection between the ICA proximal to the PCommA and the mid-basilar artery (BA). A persistent ProAtlA is very rare with fewer than 50 cases reported. Existence of a persistent carotid-vertebrobasilar anastomosis at the level of the otic vesicle, termed the otic artery, is controversial and its existence implausible according to Lasjaunias et al.34 A persistent HA arises from the C1–C3 level ICA and enters the cranium through the hypoglossal canal to form the terminal VA, giving rise to the ipsilateral PICA and BA. The VAs are hypoplastic or absent, with the contralateral VA terminating in the PICA.8 Finally, a persistent ProAtlA variant connects the ipsilateral proximal ICA or more rarely the ECA or common carotid artery to the interoccipito-atlantal horizontal VA segment, with typical hypoplasia or aplasia of the VAs. Changes in flow dynamics related to PVAs may predispose aneurysm formation or involvement with brain arteriovenous malformations (bAVMs), and these vessels can also cause neurovascular conflict. Failed regression occurs by an unknown mechanism, with flow through the primitive vasculature (purely endothelial lined channels at this stage in development) near zero at the time of resorption.8
Azygous ACA
A fused ACA, commonly involving the A2 branches, has a reported prevalence of between 0.2% and 4%.35 Increased flow through the segment increases the incidence of aneurysm formation distal to the fusion, reported in 13–71% of cases.23 An azygous ACA is also associated with holoprosencephalic failures in midline cleavage, which is different in etiology as the number of ACAs to cerebral hemispheres is matched.8
Infraoptic course of the ACA
This pattern of development occurs when the ACA originates at the level of the ophthalmic artery and courses inferior to the ON, potentially cutting through it or the optic chiasm. The development is complex and well described elsewhere.26
Persistent primary olfactory artery
The ACA rarely courses ventrally along the olfactory nerve before acutely turning back to join cortical territory distally or the anterior ethmoidal artery.8 23 Noted associations are the absence of the RAH and ACommA.
MCA duplication and accessory MCA
These variants depend upon whether both MCAs arise from the ICA (duplication) or one is accessory, arising from the ACA. Regardless, the MCA lenticulostriate arteries will arise from the most cranial of the two vessels—an anatomical feature that is of importance for risk assessment when treating intracranial atherosclerotic disease.23
Pure arterial malformations
A number of case reports have described the rare appearance of pure arterial malformations (PAMs), where overlapping dolichoectatic arteries present as a mass of loops or coiled appearance that may harbor calcifications or aneurysms.36–38 They may involve any intracranial artery, do not demonstrate arteriovenous shunting or a venous component, and seem to have a benign natural history. Reported PAMs occur more often in women, with some cases demonstrating malformations of cortical development in the vascular territory of the PAM.36 39
Fetal PCA
Fetal PCA is a misnomer given that it is actually an embryonic configuration. If the ipsilateral P1 segment is smaller in size than the PCommA or absent, the majority or all of PCA vascular territory is supplied by the carotid system. With an estimated prevalence of between 20% and 30%, unilateral or bilateral configurations are possible (approximately 10% right, 10% left, and 10% bilateral).35 40 Importantly, the size of the PCommA is unrelated to the presence of the tuberothalamic perforators, which can arise with the paramedian perforators of the ipsilateral P1 segment in 30% of cases.23
‘Duplicate’ PCAs/hyperplastic AChA
If the PCA fails to annex its common inferior temporo-occipital and medial occipital vascular territories from the AChA, the latter may appear more prominent and give an appearance of a ‘duplicated’ PCA. The reported prevalence is in the range of 1–2%.41 Duplicated AChAs have also been reported, which tend to relate to a separate origin of the hippocampal branch from the AChA.8
Incomplete fusion of the basilar tip
The basilar artery in its ‘mature’ and most common configuration (~80% of cases) is fused to the level of the pontomesencephalic sulcus with symmetric appearing PCAs. This is referred to as a cranial fusion. Two important variants exist.23 A caudal fusion results in a ‘V’-shape of the basilar tip, while an asymmetric fusion can also exist. In the case of the asymmetric configuration, it is important to note that the thalamic perforators tend to originate from the superior PCA limb in an artery of Percheron configuration, while the inferior limb is the more common site for aneurysm formation.23
Variations in SCA, AICA, and PICA
Lasjaunias understood the embryologic organization of blood supply to the brain stem and cerebellum as following that of the spinal cord, and thus comprising longitudinal anterior (BA and anterior spinal artery) and posterior (lateral medullary PICAs and posterior spinal arteries) arteries that provide perforators (basilar perforators and sulcocomissural arteries) and transverse arteries (transverse pontine arteries, coronary arteries).34 Fusion of transverse arteries reults in formation of the SCA, AICA, and distal portion of the PICA, which importantly connect through longitudinal posterior artery anastomoses.42 This elegant description provides an understanding of the formation of all observed anatomic configurations of the posterior fossa arteries, including duplications and AICA/PICA complexes.
An important concept from this model relates to extradural origins of the PICA, which has a reported prevalence of up to 20%.43 In approximately 50% of cases the lateral medullary perforators are supplied by the PICA; however, this is less likely the more lateral the origin of the vessel. Therefore, PICAs with extradural origins are less likely to harbor brainstem perforators.23 In this scenario, the brainstem perforators arise from the intradural vertebral artery.
Venous system development
During weeks 5 and 6 the vascular system is confined to the meninx primitiva. The germinal centers are first supplied by capillary loops connecting to the superficial primitive vasculature, meaning the first veins to develop are transcerebral.8 The tela choroidea develops into the choroid plexuses during weeks 6 to 8, with venous drainage initially through a ventral diencephalic vein toward the primitive transverse sinus and later through prominent bilateral dorsal choroidal veins. The main collector of the superior choroidal veins is a single, median dorsal vein, the median prosencephalic vein of Markowski (MPVM). The MPVM is not in the tela choroidea, but is a bridging vein to the dorsal dura, receiving the superior choroidal veins and draining to the interhemispheric marginal sinus (stage 5 in figure 2).7 The MPVM exists from weeks 6 to 11, when the vein of Galen (VoG) annexes the choroidal venous drainage with its inclusion into the subependymal drainage system via the developing internal cerebral veins (ICVs) that extend into the tela choroidea.44 As the venous drainage of the germinal zones, dorsal basal ganglia, and thalamus transitions between weeks 9 and 11 from transcerebral to the subependymal system, their drainage begins to outweigh the purely choroidal drainage of the MPVM leading to its regression, with the dorsal MPVM developing into the VoG and straight sinus of the deep venous system.8
The superficial venous system development is more complex, modified by the growth of the calvarium and the expansion of the cerebrum (figure 3). It starts with formation of bilateral ventral primary head veins that initially course medial to the emerging cranial nerves but caudally remodel laterally to connect with the cardinal veins.20 As they are interrupted by the otic vesicles while coursing laterally, formation of a dorsal collateral plexus is initiated, with some channels being dural while others are pial, around week 5 (5 mm).8 This venous plexus is divided into groups that drain via three stems into the primary head vein: an anterior plexus covering the forebrain and midbrain (stem at the trigeminal ganglion), a middle plexus covering the metencephalon (stem between the trigeminal ganglion and otic vesicle), and a posterior plexus covering the myelencephalon (stem along the vagus nerve).23 The primitive maxillary vein is the most cranial tributary of the primary head vein and drains the optic vesicle. The primary head vein cranially remains medial to the trigeminal ganglion where the cavernous sinus will later form.20
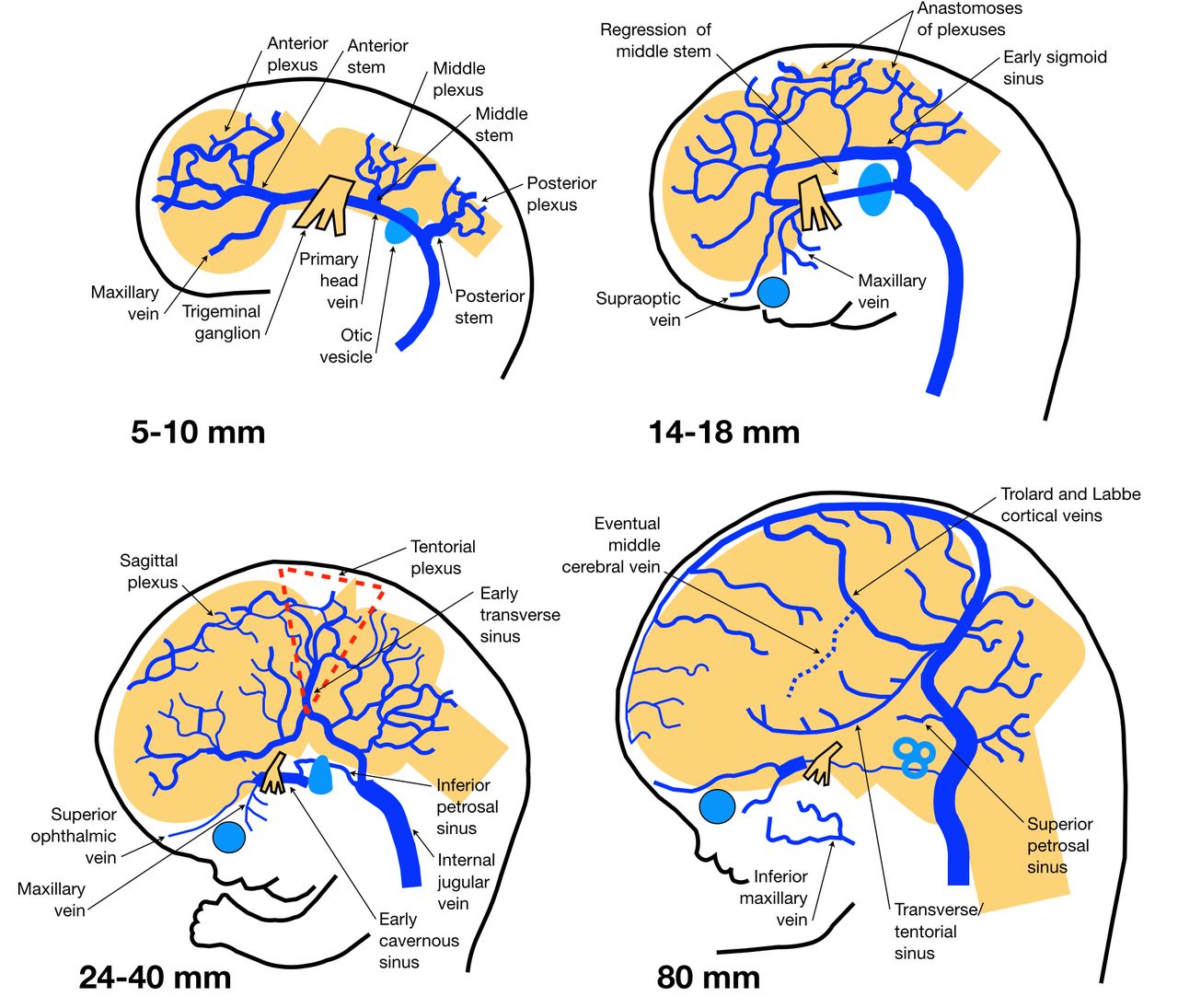
Development of the superficial cerebral venous system. Sizes provided refer to crown–rump length. See text for details. Adapted from Mullan et al,20 with permission.
By week 6 (10 mm), the anterior venous plexus anastomoses dorsally with the middle plexus, which will eventually contribute to the transverse sinus (TS). As the primary head vein reorganizes from medial to lateral of the vagus nerve through collaterals, a medial remnant becomes the inferior petrosal sinus while the cardinal vein will develop into the internal jugular vein. By the seventh week (10–16 mm) the ventral surface of the brain demonstrates conspicuous dural and pial arachnoid venous channels in response to the vascularization of the germinal matrix and striatum. During late week 7/early week 8 (16–21 mm) the choroid plexuses begin to develop, with the forebrain plexus drained by a ventral pial diencephalic vein.8 As the otocyst enlarges (previously the otic vesicle), the middle and posterior plexuses form a dural anastomosis with the posterior stem that becomes the sigmoid sinus.23 With enlargement of the cerebral hemispheres, flow starts to direct preferentially to regions where the superficial plexuses are protected, such as between the hemispheres and the calvarium in the case of the superior sagittal sinus, between the cerebral hemispheres, calvarium, and cerebellum to form the TS, and between the cerebral hemispheres and tentorium for the straight sinus.8 This expansion also stretches the cortical veins into a lateral orientation.20 The dural networks overlying the dorsal prosencephalon and mesencephalon are brought into apposition and fuse, producing the tentorial plexus. Plexuses in the falx and tentorium usually persist during fetal life.8
By week 8 (18–26 mm) the ‘choroid stage’ is present and the MPVM drains posteriorly into the tentorial plexus as described above. The otocyst further enlarges to obliterate the remainder of the primary head vein, directing all drainage into the posterior dural network. The middle plexus and primary head vein form the pro-otic sinus anterior to the otocyst and connect with the primitive maxillary vein (which is incorporated into the external jugular vein with the inferior ophthalmic vein remnant) and primitive supraorbital vein (which later forms the superior ophthalmic vein), forming the primitive cavernous sinus.20 While the TS expands and the middle cerebral vein becomes more prominent, the anterior stem regresses to potentially form the uncal vein.1 7
By week 9 (40 mm) the superior sagittal sinus is well developed and the tentorial plexus elongates and aligns parallel to the TS.8 Chondrification of the occipital bone embeds the condylar, mastoid, and occipital emissary veins. By week 12 (60–80 mm) the torcula has a variable plexular appearance and the middle cerebral vein is pulled anterior to the edge of the lesser sphenoid wing by the enlarging cerebral hemispheres. The ICVs are fully formed, connecting dorsally with the basal veins of Rosenthal, which links telencephalic, diencephalic, and mesencephalic veins, providing multiple redundant drainage routes.
Congenital arteriovenous lesions
There are three reported types of congenital arteriovenous shunting lesions: vein of Galen malformations (VoGMs), pial fistulas, and dural sinus malformations (DSMs).45 Genetic testing has recently helped further understand mechanisms behind the development of these lesions. Owing to their incomplete penetrance, Lasjaunias proposed a two-‘hit’ model to cause the arteriovenous shunting lesion phenotype.46 In this model, the first ‘hit’ is a germline mutation, while the second ‘hit’ involves application of a revealing trigger, most commonly thought to be ischemic or inflammatory, to cause a somatic mutation that unmasks the vascular defect.47 48 Timing, target, and location of the revealing trigger are essential in determining the type of lesion that forms. This model has been confirmed for the development of arteriovenous shunting lesions in hereditary hemorrhagic telangiectasia (HHT) and capillary malformation-arteriovenous malformation (CM-AVM) syndromes.49–51
Development of congenital arteriovenous shunting lesions has previously been understood to occur due to fistulization after intrauterine thrombosis, which is also thought to form benign developmental venous anomalies.20 52–55 However, with the insights from genetic testing, it is now more reasonable to speculate that lesion formation is secondary to dysfunction in normal angiogenesis. Since the primitive network of vessels in the meninx primitiva remains plastic in regards to arteriovenous specification during vasculogenesis, it is unlikely for a fistula to form at this stage of neurovascular development. For much of in utero development the neurovasculature is composed of simple endothelial lined tubes, with a muscularis layer not forming until ~20 weeks for striatal arteries and not until the end of term for extrastriatal arteries.8 Full function of the cerebral blood vessels does not occur until after birth, with neurovascular coupling developing during the first few weeks of postnatal life.56 In many animal models erroneous vasculogenesis is fatal, while several of the genes associated with these lesions (HHT genes, for instance) are not expressed until later in development.57 Finally, analogous arteriovenous shunting lesions (bAVMs and dural arteriovenous fistulas) arise during adulthood, when vasculogenesis can no longer occur. Therefore, its is most reasonable to consider dysfunctional angiogenesis at the capillary level joining arteries to veins, starting from the deep pial layer of the primitive vascular plexus, as the mechanism and site for arteriovenous fistula creation.
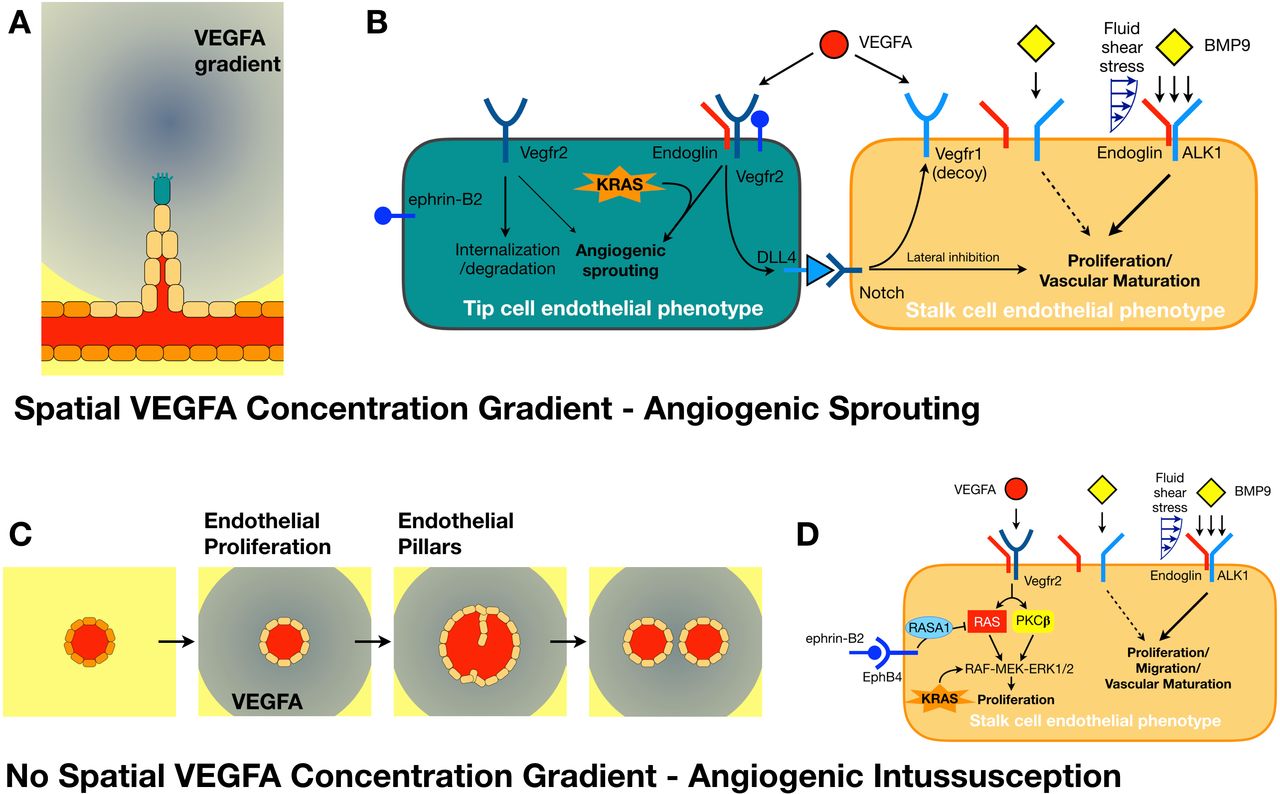
As there are many genes and their products involved in angiogenesis with overlapping functions, to conceptually simplify their significance, they may be grouped according to whether a gain-of-function or loss-of-function mutation affects (a) TC or SC phenotype, or (b) intussusceptive angiogenesis (figure 4). Sprouting angiogenesis occurs in response to a VEGFA concentration gradient and requires a balance between the number of TCs and SCs to produce a capillary bed with a normal hexagonal pattern and hemodynamic impedance (figure 4A).18 19 58 Angiogenesis in tumors is not as well coordinated and therapy that inhibits SC formation (through blocking Notch signaling) alters the TC:SC ratio to produce a hypersprouting vascular bed that is leaky and ischemic.59 60 This ischemia may provide a revealing trigger for focal EC somatic mutations that weaken the vessel and cause it to dilate in response, forming a fistula.61 A fistulizing dilatation may also result from dysfunctional angiogenic intussusception. Without a spacial gradient in VEGFA concentration, it is suspected that ECs all convert to SCs, and as part of the phenotype, proliferate.58 As the vessel dilates, intraluminal endothelial pillars form (thought to arise from adhesion of circulating endothelial progenitor cells) and fuse to divide the vessel (figure 4C).17 However, if the rate of EC proliferation causes a vessel to dilate too fast, the pillars cannot fuse and an enlarged capillary, or fistula, results.62

Extravascular sprouting and intravascular intussusceptive angiogenesis. (A) A spatial gradient in vascular endothelial growth factor A (VEGFA) from ischemic tissue results in endothelial cell (EC) phenotype change of quiescent phalanx cells (dark orange) into a tip cell (TC, teal) followed by a cord of stalk cells (SCs, beige). The TC uses filopodia to navigate and migrate into the ischemic tissue. (B) A simplified perspective associating different signaling molecules with a TC or SC phenotype in sprouting angiogenesis. (C) In the setting of a uniform abluminal VEGFA concentration, all ECs are thought to attain a SC phenotype and proliferate. Endothelial pillars form that can divide the vessel through intussusception to regulate tissue perfusion. (D) Simplified perspective of the different signaling molecules involved with the SC phenotype in intussusceptive angiogenesis.
A hypersprouting vascular bed may occur if a mutation causes increased TC phenotype or decreased SC phenotype in the presence of a VEGFA concentration gradient. Examples of the former include gain-of-function mutations in oncogene KRAS demonstrated mosaically in brain AVMs63 64 and ephrin-B2, which has been shown to be increased in pedatric bAVMs and assists with VEGF-endocytosis related signaling.65–68 Decreased SC phenotype is typically associated with mutations that define HHT, with reduction in BMP9 (or BMP10 in utero) related transforming growth factor signaling through the ALK1 pathway. Approximately 85–90% of HHT cases demonstrate mutations in ALK1 (HHT subtype 2) or its coreceptor endoglin (HHT subtype 1).48 69 Blood flow related mechanotransduction plays an important role in some of these pathways, with shear stress regulating ALK1 signaling potentiation by endoglin (and proposed as the etiology for formation of malformations of cortical development seen in 10–15% of patients with HHT),48 61 while ephrin-B2 expression is dynamic in response to shear stress.70 This hypersprouting probably contributes to the increased blood–brain barrier permeability of bAVMs.64 71
Abnormal vessel dilatation with defective intussusception has been recently demonstrated for loss of function mutations in EphB4.62 It was shown that EphB4 plays a key role, decreasing VEGFA signaling by up to 40% to slow SC proliferation and allow intussusception to occur, independently of the BMP9/ALK1 and angiopoietin/Tie2 pathways. VEGFA signaling induces SC proliferation by two pathways (RAS and PKC) that converge on the RAF-MEK-ERK1/2 cascade (figure 4D).72 Normally, EphB4 inhibits the RAS pathway to slow proliferation and allow for intussusception without arresting vascular development.62 Noting RASA1 is downstream of EphB4 along this pathway, these mutations probably also produce arteriovenous shunts through failed angiogenic intussusception.73 74 Platelet-derived growth factor BB seems to have a similar role, regulating angiogenic intussusception where related mural cell recruitment limits EC proliferation; however, this vascular maturation, which includes organizing the glia limitans of the glymphatic system, does not seem to occur until postnatally.75 76 Interestingly, KRAS gain-of-function mutations cause increased MEK-ERK1/2 signaling,63 64 whereas endoglin loss-of-function mutations independently cause abnormal vessel growth with loss of normal EC migration,77 implicating overlap of hypersprouting and excessive SC proliferation mechanisms for lesion formation.
This approach helps to simplify the complex information provided by genomic analysis studies and to better understand the underlying pathology of the lesions. It should be noted that this field is advancing rapidly, with even differences in endothelial function and angiogenesis now demonstrated by biological sex.78
Vein of Galen malformations
VoGMs are purely choroidal malformations and account for approximately 30% of pediatric arteriovenous shunts.79 The location of the lesion is extraparenchymal, within the meningeal space of the velum interpositum and ambient cisterns.8 The primary arterial feeders are the choroidal and transmesencephalic collicular arteries to the tela choroidea and quadrigeminal plate, respectively, with recruitment of secondary arterial feeders later in fetal and postnatal life.80 Venous drainage is via a persistent, midline dilated MPVM, which can present with a falcine sinus (figure 5), absent straight sinus, or ‘falcine loop’ as described by Raybaud.7 These venous outflow variations, although characteristic, are also seen in ~2% of the population.81 A very important consideration is the presence of deep venous drainage into the VoGM via the ICVs, first described by Raybaud and stressed by Gailloud and colleagues.7 82 These may not be visible in a high-flow fistula without ICV reflux, but are present in a third of cases with occlusion of the ICVs during endovascular treatment the most significant cause of iatrogenic morbidity and mortality.83–85

Vein of Galen malformation (VoGM). (A) Midline sagittal gray scale and (B) colour Doppler ultrasound of an infant boy presenting with cardiorespiratory insufficiency. The hypoechoic venous pouch of the VoGM (white arrow) demonstrated high-flow arteriovenous shunting with pulse wave Doppler (not shown). (C) Midline sagittal time of flight MR angiogram demonstrates multiple superior choroidal arterial feeders from the anterior cerebral arteries (white arrowhead) with arterialization of the VoGM pouch (white arrow) and a falcine dural sinus (white*). Left internal carotid artery digital subtraction angiogram in the anterior posterior (D, E) and lateral (F, G) projections during the early (D, F) and late (E, G) arterial phases of the same patient demonstrate numerous superior and anterior choroidal arterial feeders into the VoGM pouch that drains into the falcine sinus.
Recent gene sequencing has reported ~10% of patients with VoGMs have an EphB4 germline mutation, while exome sequencing found that mutations in chromatin modifier genes and EphB4 accounted for ~30% of cases.50 86 Although lesion development was previously thought to relate to thrombosis of the straight sinus, VoGMs may instead be related to failed angiogenic intussusception during development of the choroid plexuses, triggered by an ischemic event. In the presence of an arteriovenous shunt, transport of VEGFA and other related growth factors downstream is enhanced and may be responsible for the failed regression of the MPVM.87 Although VoGMs are rare and appear sporadic, patients with CM-AVM syndromes I and II can present with VoGMs along with intracranial and extracranial AVMs and multifocal capillary malformations and telangiectasias, with this mechanism providing a plausible explanation for co-existence of all these lesions.88–90
Pial fistulas
Reports suggest that more than half of pediatric pial fistulas involve RASA1, HHT1, or HHT2 germline mutations.89 Following the revealing trigger, fistula progression to a bAVM has been suggested to occur through retention of downstream vascular channels due to the increased flow-related shear stress that also induces arteriogenesis (arterialization of a capillary).61 91 This forms the bAVM nidus, with this sequence of lesion progression demonstrated in patients with HHT.92 As cerebral blood flow is very low during early development and increases threefold in the third trimester and 1.5-fold in the first 2 weeks of life, these flow-related bAVM maturation events are unlikely to occur in utero.93–95 If the timing of the revealing trigger is very early, prior to the migration of the neural crest, a metameric distribution of arteriovenous lesions may present in keeping with cerebrofacial arteriovenous metameric syndrome.96 97
RASA1 mutations are classically associated with CM-AVM type I and associated high-flow pial fistulas and capillary malformations (figure 6A–G).98 Interestingly, ephrin-B2 is detected in the urine of pediatric patients with bAVMs, questioning whether a significant mode of vascular ephrin-B2-EphB4 signaling occurs through shearing of ephrin-B2 ligands from arteries into the blood and binding downstream to venous EphB4 receptors.68

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pial fistula and torcular dural sinus malformations (tDSMs). (A) A young pediatric patient with a RASA1 germline mutation presented with decreased level of consciousness and was found to have a left medulla parenchymal hematoma with surrounding edema on non-contrast CT. (B) The hematoma was surrounding two arterialized venous pouches with coronal CT angiography images. Surrounding brainstem edema is seen on (C) coronal T2-weighted and (D) axial fluid-attenuated inversion recovery (FLAIR) images. Anteroposterior (E) and lateral (F) digital subtraction angiograms demonstrate a single hole pial arteriovenous fistula from the lateral medullary segment of the right pica (**) draining through a transosseous vein into the right internal jugular vein (white arrow). There are bilobed post-fistulous venous pouches (black arrow). A second single hole pial arteriovenous fistula (*) also arises from a hypertrophied medullary perforator, shunting into the deep venous system via lateral mesencephalic veins (black arrowhead) and into the clival epidural plexus (white arrowhead). (G) Anteroposterior right vertebral artery angiogram following successful treatment using coil and glue embolization of both pial fistulae. (H) A large tDSM in a patient is demonstrated by a large hypointense dural venous lake with mass effect on the adjacent brain on T2-weighted fetal MR images. (I) Phase encoded artifact of the tDSM is noted on gradient echo MR images related to the pulsatile arterialization of the venous sinus. Anteroposterior (J) and lateral (K) digital subtraction angiograms following a left common carotid artery injection demonstrate a high-flow jet of contrast from a posterior branch of the middle meningeal artery feeder into the enlarged dural venous sac (black arrow heads). (L) Sagittal T1-weighted and (M) axial FLAIR images of a similar lesion in a newborn patient who underwent staged endovascular embolization of the arterial feeders. (N) Axial T2-weighted MR and (O) coronal maximal intensity projection time of flight venogram images 14 years later demonstrate normal developmental appearance of the brain and remodeling of the dural sinuses.
Dural sinus malformations
With advances in obstetric imaging, thrombus formation in the dural sinuses causing enlargement of the torcula is more common than previously thought, and fortunately, now known to spontaneously regress with good neurological outcome in approximately 70% of cases.99 100 If arteriovenous fistulization occurs, patients can present with significant neurological symptoms as the shunt competes with the normal venous drainage pathway for the neonatal brain via the dural sinuses. This effect is much more dramatic than in adults as venous drainage of the middle cerebral vein through the cavernous sinus does not occur until after birth, meaning there is no compensatory collateral venous drainage for the neonatal brain.20 Abnormal remodeling of the jugular bulb in response to the fistula also necessitates early treatment.101 The dural fistulas are often bilateral with worse prognosis if there is coexisting cerebral ischemia, hydrovenous disorders, and/or cardiopulmonary insufficiency.102 These shunts may involve the torcula or sigmoid sinus and classically present with the clinical triad of macrocrania, cranial bruit, and signs of cardiac failure.103 The best treatment is staged embolization of arterial feeders until flow reduction enables a natural regression of the lesion in combination with anticoagulation (figure 6H–O).
Although mutations involving RASA1 and PTEN have been reported for congenital DSMs,103 a mechanism similar to that suggested in adults seems most appropriate. This stems from a fistula forming in the developing vasa vasorum of the dural sinuses.104 Recently, it was demonstrated that low endothelial expression of connexin37 proteins, which are downstream of ALK1 signaling, produces vessel dilatation and arteriovenous shunts.105 This expression was shown to plummet in the presence of tumor necrosis factor, suggesting inflammation related to the thrombosis may contribute to fistula formation. Significant increases in acute phase reactants have been noted in patients with DSMs with the onset of thrombosis of their lesions.101
Conclusions
Having an understanding of cerebral neurovascular development is important to fully appreciate the implications of anatomic variants and congenital arteriovenous lesions requiring treatment encountered in endovascular neuroradiological practice. Identifying such variations in the neurovasculature may also be an indicator of an adjacent structural malformation. As the interplay between hemodynamics and genetics is being more thoroughly understood, new insights into mechanisms of arteriovenous fistula formation are being discovered. At present, angiogenic disruptions resulting in hypersprouting and accelerated endothelial proliferation seem the most likely mechanisms for development of congenital arteriovenous shunting lesions.
Supplemental material
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
Acknowledgments
JK receives career scholarship financial support from the Fonds de recherché du Quebec-Sante (FRQS).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Both authors contributed to the ideas and writing of the manuscript, its review, and approved the final version. The manuscript was also kindly reviewed by Professor Mohamed Aggour and Professor Jens Fielher on behalf of the European Society of Minimally Invasive Neurological Therapy (ESMINT) - Basic Science Education Initiative.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests TK: royalties from Thieme; stock/stock options from Marblehead; paid consultant for Medtronic, Stryker, Penumbra, and Cerenovus.
Provenance and peer review Commissioned; internally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.