Article Text

Abstract
Background and purpose The recent randomized trials demonstrated the benefit of mechanical thrombectomy in stroke therapy. However, treatment using different strategies is an ongoing area of investigation. The PROMISE study analyzed the safety and effectiveness of the Penumbra System with the ACE68 and ACE64 reperfusion catheters in aspiration thrombectomy of stroke, using A Direct Aspiration First Pass Technique (ADAPT).
Methods PROMISE was a prospective study which enrolled 204 patients with intracranial anterior circulation large vessel occlusion (LVO) ischemic stroke in 20 centers from February 2016 to May 2017. Initial treatment was with the ACE68/ACE64 catheters within 6 hours of symptom onset. Imaging and safety review was performed by an independent Core Laboratory and a Clinical Events Committee. The primary angiographic outcome was revascularization to mTICI 2b-3 at immediate post-procedure and the primary clinical outcome was 90-day modified Rankin Scale (mRS) score ≤2. Safety assessment included device- and procedure-related serious adverse events (SAEs), symptomatic intracranial hemorrhage (sICH), mortality, and embolization of new territory (ENT).
Results Enrolled patients had a median age of 74 (IQR 65–80) years and a median admission NIHSS of 16 (IQR 11–20). The post-procedure mTICI 2b-3 revascularization rate was 93.1% and the 90-day mRS 0–2 rate was 61%. Device- and procedure-related SAEs at 24 hours occurred in 1.5% and 3.4%, respectively, 90-day mortality was 7.5%, sICH occurred in 2.9% while ENT occurred in 1.5%.
Conclusions For frontline therapy of LVO stroke, the ACE68/ACE64 catheters for aspiration thrombectomy were found to be safe and showed similar efficacy to randomized trials using other revascularization techniques.
Clinical Trial Registration NCT02678169; Pre-results.
- stroke
- thrombectomy
- device
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Six randomized controlled trials (RCTs) established the role of mechanical thrombectomy in early acute ischemic stroke (AIS):1–6 in these trials, most of the devices used were stent retrievers.7 As a result of position statements recommending endovascular therapy,8–10 a significant growth in the treatment of patients with emergent large vessel occlusion (ELVO) has been observed and is expected to continue.11–13 An operational definition of ELVO to standardize the description of the therapeutic target for endovascular thrombectomy was proposed.14 Recently, several studies have investigated patient outcomes after advances in different devices and techniques, increased physician experience, and streamlined in-hospital procedures for endovascular therapy.15–18 The Penumbra System (Penumbra, Inc., Alameda, CA) is a family of mechanical thrombectomy devices specifically designed to remove thrombus through aspiration, receiving 510(k) clearance from the US FDA in 2007.19 20 THERAPY was an RCT assessing aspiration thrombectomy with IV rt-PA versus IV rt-PA alone.21 Further developments included ADAPT (A Direct Aspiration First Pass Technique), a thrombectomy approach wherein aspiration alone is used to remove the occlusion, followed by adjunctive therapies if necessary.22–24 In addition, findings from the Penumbra 3D RCT, which randomized patients to either a frontline approach with the 3D stent retriever with aspiration vs aspiration alone, demonstrated no significant difference between treatment groups in the rates of modified Thrombolysis in Cerebral Infarction (mTICI) 2–3 revascularization, 90-day modified Rankin Scale (mRS) 0–2, symptomatic intracranial hemorrhage (sICH), procedure or device-related serious adverse events (SAEs), or mortality.25
At the time of this study’s design, the safety and efficacy of the ADAPT technique using ACE68/64 catheters with the Penumbra System (PS) had not been assessed in a prospective study. The aim of a prospective, multicenter, observational, single-arm European Registry on the ACE reperfusion catheters and the Penumbra System in the treatment of acute ischemic stroke (PROMISE) study was to address this need for real-world data on the safety and effectiveness of the PS with the novel Penumbra reperfusion catheters, ACE68/64, in patients with AIS from anterior circulation LVO, treated with the ADAPT technique. The PROMISE study incorporated an integrated aspiration thrombectomy system which includes a Penumbra aspiration pump indicated for revascularization of AIS patients.
Methods
PROMISE was a prospective, multicenter, single-arm, open-label, observational European study to evaluate the safety and effectiveness of the ACE68/64 reperfusion catheters and the PS in the treatment of AIS (ClinicalTrials.gov Identifier, NCT02678169). Patients presenting within 6 hours from symptom onset with an anterior circulation LVO AIS (within the internal carotid artery (ICA) and internal carotid terminus, middle cerebral artery (MCA) – M1/M2 segments) were eligible. This study was a non-inferiority comparison of 90-day mRS 0–2 rates between the ACE Penumbra System group and the intra-arterial therapy (IAT) group in the MR CLEAN trial.1 At the time of study design, MR CLEAN established the benefit of intra-arterial therapy against medical management alone and was the only published RCT for our non-inferiority hypothesis and sample size calculation. The main aim of the study, however, was to assess the safety and effectiveness of the ACE68/64 in patients treated with the ADAPT technique. Every patient was checked for study eligibility and recorded on a site-specific Screening Log.
Treatment consisted of the ADAPT technique with thrombo-aspiration using ACE68 and ACE64 as first intention according to site routine practice. For rescue, any approved revascularization device and/or intra-arterial medication was allowed at the operators’ discretion, including non-Penumbra devices. Adjunctive therapy was defined as the use of a stent retriever following a reperfusion catheter: before adjunctive therapy, three passes of the ACE catheter were recommended. At the discretion of the clinicians in charge of the patient or the investigator, the patient was pretreated with intravenous thrombolytics (IV tPA), and/or treated under general or local anesthesia, or conscious sedation. Follow-up visits to assess functional outcome, quality of life, and adverse events took place at 24 hours, 7 to 10 days or discharge, 30-days, and 90-days post-procedure. In Europe, the ACE68 and ACE64 catheters were available in 2016 and 2015, respectively.
All procedures were in accordance with national and local ethical and institutional guidelines and site routine clinical practice. The local Ethics Committees approved the study, and the patients or their representatives provided written informed consent, according to local regulations.
Imaging analysis
All imaging scans (baseline CT/MRI, pre-procedure/procedure DSA, and 24 hours' scan) were reviewed and adjudicated by an independent Core Imaging Laboratory (Toronto Western Hospital, University of Toronto) for mTICI and for procedural and safety assessments (confirmation of vessel damage and embolization of new territory (ENT), identification, and classification of intracranial hemorrhages).
Patients
Patient eligibility criteria included age of at least 18 years, presentation with anterior circulation LVO within 6 hours of stroke onset or time last known to be well, occlusion of the internal carotid artery (ICA and ICA terminus) or MCA M1/M2, National Institutes of Health Stroke Scale (NIHSS)≥2, and pre-stroke mRS score ≤2. LVO status was determined mostly by CTA (85.3%), with some by MRA (14.7%). Imaging eligibility criteria were CT Alberta Stroke Program Early CT Score (ASPECTS)≥6 or MR DWI ASPECTS≥5, and absence of tandem extracranial occlusion or severe arterial stenosis requiring treatment prior to thrombectomy. Study exclusion criteria can be found in the Online Supplement. Up to 210 patients who met eligibility criteria were to be enrolled in a maximum of 25 European investigational sites, and an exclusion log of screened but not enrolled patients was kept at each site.
Primary and secondary endpoints
Primary endpoints were angiographic revascularization assessed by digital subtraction angiography of the occluded target vessel to mTICI 2b or 3 at immediate post-procedure as adjudicated by an independent Core Laboratory and functional independence (mRS 0–2) at 90 days by mRS certified medical personnel, without blinding to the clinical or radiological patient data. Secondary endpoints were any device- and procedure-related SAEs at 24 hours' and 30 days' post-procedure, all-cause morbidity (defined as mRS 3–5) and mortality at 90 days, occurrence of ENT, occurrence of sICH at 24 hours, and occurrence of vessel damage at the end of the ADAPT procedure. Safety was assessed by collecting adverse events data during all study visits. Hemorrhagic transformation was radiologically classified according to the European Cooperative Acute Stroke Study (ECASS) definitions. sICH was defined according to ECASS-II definition26 27 as a four point or greater deterioration in the NIHSS within 24 hours after treatment compared with the pre-procedure NIHSS attributable to imaging evidence of an intracranial hemorrhage as adjudicated by the Core Imaging Laboratory. Safety endpoints were adjudicated for severity and causality by an independent Clinical Events Committee (CEC). All data were monitored during on-site visits.
Secondary efficacy endpoints included good functional neurological recovery defined as a reduction of 10 or more points in the NIHSS or a score of 0–1 at 7 to 10 days' post-procedure, times to mTICI ≥2 b revascularization, health economics data, and Quality of Life as assessed by EQ-5D-3L score at 90 days compared with 7 to 10 days.
Statistical analysis
Baseline clinical and imaging data were summarized using standard descriptive statistics. This included the number of observations, mean, median, SD, minimum and maximum for continuous variables, and counts and percentages for discrete variables. All confidence intervals presented were two-sided. All statistical tests were two-tailed with a significance level of 0.05. Analyses were performed using SAS software (version 9.4; SAS Institute).28
The primary effectiveness analysis was an unadjusted non-inferiority comparison between the ACE68/64 reperfusion catheters Penumbra System group and the IAT group from the MR CLEAN trial. For sample size calculation, it was assumed that 38% (76/200) of the PS patients would experience primary endpoint success for 90-day mRS 0–2 compared with 32.6% (76/233) of the MR CLEAN IAT group: the estimated difference and 95% CI for the difference between groups would therefore be 5.4% (-3.7%, 14.4%). As the primary analysis, all efficacy and safety outcome measures were analyzed under the intent-to-treat (ITT) principle.
Prespecified analyses of association between primary and selected secondary endpoints (mTICI 2b-3 post-procedure, 90-day mRS 0–2, device- or procedure-related SAEs at 24 hours and 30 days, 90-day mortality, occurrence of sICH) were conducted, with adjustment for several baseline characteristics (age, baseline NIHSS, occlusion location).
Results
Baseline patient data
Between February 2016 and May 2017, the PROMISE study enrolled 204 patients across 20 European centers using ADAPT with the Penumbra System and ACE68/64 catheters as frontline treatment. For enrollment, 30 (14.7%) patients were selected by MRI and 174 (85.3%) by CT. Of the 1433 patients screened and documented in site-specific Screening and Enrollment Logs, 1229 patients (85.8%) were screen failures and 204 patients (14.2%) were enrolled. The five most common reasons for screen failure were no imaging evidence of LVO of the anterior circulation (22.1%, 271/1229), followed by unknown onset of stroke symptoms with last proof of good health more than 6 hours before (10.3%, 126/1229), lack of possibility to obtain informed consent (10.2%, 125/1229), onset of stroke symptoms >6 hours (8.2%, 101/1229) or angiographic evidence of tandem extracranial occlusion or arterial stenosis proximal to occlusion requiring treatment prior to thrombus removal (8.2%, 101/1229). Four patients withdrew consent prior to the 90-day visit, therefore, 200 patients completed the 90-day visit and were included in the follow-up analysis.
Baseline characteristics are displayed in table 1. The median age was 74 years, ranging from 27 to 96 years, and almost two-thirds of the patients were women (61.8%). Thirty-five percent (35.3%) of patients were transferred from other hospitals, 79.9% of stroke onsets were witnessed, and 20.1% of patients were last known to be well within 6 hours before inclusion without knowing the exact time of onset. The median time from stroke onset to hospital admission was 97 min (IQR 58.5–154.5). The median baseline NIHSS score was 16 (IQR 11–20) and ranged from 2 to 36. At admission, the median CT ASPECTS was 9, while the median MRI DWI ASPECTS was 8. Occlusions were located in the ICA/carotid T (21.1%) and MCA (78.9%; 60.8% in M1, 18.1% in M2) and IV rt-PA was administered in 126 patients (61.8%), mainly within a bridging protocol 86.5% (109/126). At baseline, mTICI was 0 in most patients (91.2%, 186/204) with mTICI 1 in 8.3% (17/204) of patients.
Baseline and procedural characteristics
The ACE68 catheter was utilized as frontline treatment in 65.7% of patients and the ACE64 in 32.8%: three patients (1.5%) were initially treated with other PS devices (table 1). For cases that required adjunctive therapy, the median number of ACE68/64 passes performed before switching was 2 (IQR 1–3). General anesthesia was performed in 58.3%, conscious sedation in 18.1%, and local anesthesia only in 23.5% of patients. A stent was placed for proximal stenosis or dissection in 2% of patients, and balloon angioplasty was used for proximal stenosis in 2% of patients. In 20.9% of patients, a stent retriever was used after PS as adjunctive treatment, in 13.4% of cases at the target vessel, 9% in distal vasculature, and 1.5% in both. No balloon guide catheters were reported to be used.
Primary outcomes
The proportion of patients with Core Laboratory-assessed mTICI 2b-3 was 93.1% (190/204) after all treatment (39.2% mTICI 3, 53.9% mTICI 2b). After Penumbra System treatment alone, 70.6% (144/204) of patients achieved mTICI 2b-3. Out of all patients achieving mTICI 2b-3 after ACE68/64, 74.4% (90/121) achieved revascularization after the first ACE68/64 pass. At 90 days, 61% of patients (122/200) achieved good functional outcome (mRS 0–2) (table 2). The study met its primary effectiveness endpoint (Online Supplement).
Primary and secondary endpoints
Secondary outcomes
Regarding safety endpoints, the all-cause mortality and morbidity rates at 90 days were 7.5% (15/200) and 31.5% (63/200), respectively, and the 24-hours sICH rate was 2.9% (6/204). According to MedDRA preferred terms, three patients died due to progression of stroke, two patients died due to cerebrovascular accident, and the remaining 10 patients died due to different causes (cerebral hemorrhage, hemorrhagic transformation stroke, ischemic stroke, seven others not related to stroke). The ENT rate was 1.5% (3/204), vessel perforation rate was 0.5% (1/204), distal emboli rate was 1.0% (2/204), and vessel dissection rate was 2.5% (5/204) (table 2). The frequency of new ischemic stroke within 24 hours was 0%.
Per adjudication by the CEC, there were nine device- and procedure-related SAEs (4.4%) at 30 days, of which seven (3.4%) occurred within 24 hours. These events included carotid artery dissection (n=2), cerebral artery embolism (n=2), cerebral artery occlusion (n=1), cerebral hematoma (n=1), hemorrhagic cerebral infarction (n=1), subarachnoid hemorrhage (n=1), and vascular pseudoaneurysm (n=1). Regarding vessel damage events (serious and non-serious), there were five arterial dissections, all of which were extracranial, and one perforation during stent retriever use in distal vasculature. Four of the five dissections were unrelated to ACE catheter use and possibly/probably/definitely related to the procedure.
Regarding secondary efficacy endpoints, good functional neurological recovery was achieved in 67.9% of patients (127/187), with 35.3% (66/187) having NIHSS 0–1. The times to revascularization endpoint data are shown in table 2. The median reperfusion time (arterial access to mTICI 2b-3 or final angiogram if 2b-3 was not achieved) was 31 min (IQR 20–53), and the median time from stroke onset to mTICI 2b-3 or final angiogram was 245.5 min (IQR 192–305). The current time metrics are shown in figure 1. Please see Online Supplement for details of safety events, primary effectiveness analysis, and additional process times.

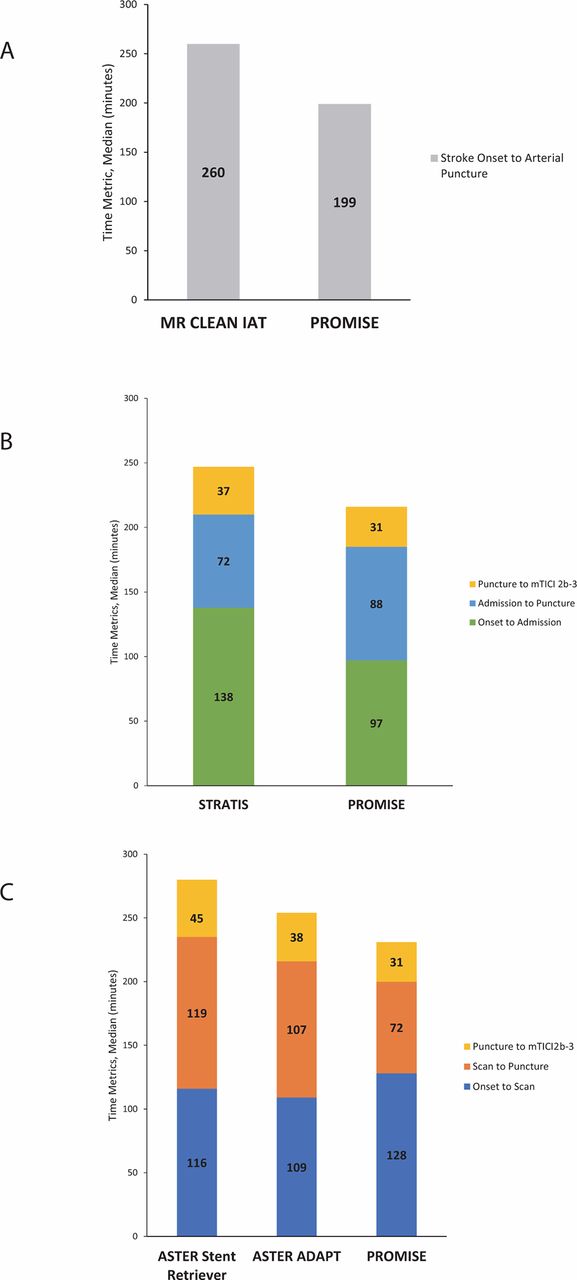{kind=link}
Comparison of PROMISE time metrics with MR CLEAN, STRATIS, and ASTER trials (A) MR CLEAN and PROMISE procedural time comparison. (B) STRATIS and PROMISE procedural time comparison. (C) ASTER and PROMISE procedural time comparison.
Self-reported assessment on quality of life at 90 days compared with 7 to 10 days included improvement in mobility (9.9%), self-care (14.5%), usual activities (19.0%), and pain/discomfort (7.1%), and worsening in anxiety/depression (4.2%). Median duration of index hospitalization was 8 days (IQR 5–11), which is typical per European standard of care, and all-cause rehospitalization rate from discharge to 90 days was 8.3% (17/204). Of note, 73.5% of patients (150/204) were back home by 90 days.
Subgroup analyses
In pre-specified subgroup analyses of primary and selected secondary endpoints, several baseline covariates were examined including stroke severity per NIHSS (<16, 16–20, or >20), age (<80 or≥80 years), and occlusion location. In unadjusted analysis, lower baseline NIHSS and younger age (<80 years) were predictors of functional independence. In addition, younger age (<80 years) was a predictor of decreased mortality. No impact of higher baseline NIHSS on the occurrence of sICH or mortality was observed. In analysis adjusted for the significant predictors of baseline or outcome differences, there were no differences in rates of 90-day mRS 0–2, mTICI 2b-3, or mortality between transfer and direct-admit patients. For subgroup analysis details, please see Online Supplement.
Discussion
Results from the PROMISE study supported the safety and effectiveness of the ACE68 and ACE64 reperfusion catheters with the PS in patients with AIS secondary to anterior LVO, using the ADAPT technique as frontline therapy in real-world practice. mTICI 2b-3 post-procedure was achieved in 93.1% of cases (including 20.9% of patients who were also treated with stent-retrievers), and 61% of cases achieved 90-day mRS 0–2. The median time from arterial puncture to mTICI 2b-3 was 31 min. The safety profile was favorable with 7.5% early mortality, 2.9% sICH, and 1.5% ENT rates.
In the ASTER RCT which randomly assigned patients to a frontline approach with contact aspiration or stent retriever, therapy with the ADAPT technique was not superior to stent retrievers in safety or efficacy.16 A recent meta-analysis (HERMES) pooled patient-level data from five RCTs comparing endovascular thrombectomy to standard medical care in patients with AIS. It showed the benefit of mechanical thrombectomy devices over medical management alone.7 The STRATIS study determined that outcomes observed in a meta-analysis (SEER Collaboration) pooling patient-level data from four RCTs could be reproduced in a large real-world cohort.15
Data from the current study were compared with published literature. Compared with ASTER, mTICI 2b-3 was achieved in 93.1% of patients in PROMISE (20.9% stent retriever use) vs 85.4% in ASTER (32.8% stent retriever use). Compared with the endovascular treatment groups in HERMES, STRATIS, and ASTER, the proportion of patients with 90-day mRS scores 0–2 was higher in PROMISE (61% vs 46% in HERMES, 56.5% in STRATIS, and 45.3% in ASTER aspiration). Age and baseline NIHSS in these trials were comparable to the PROMISE population, although other characteristics, such as tandem occlusion, may vary. The 90-day mortality rate was satisfactory in PROMISE (7.5% vs 15.3% in HERMES, 14.4% in STRATIS, 19.3% in ASTER aspiration). The rate of sICH in PROMISE was lower than HERMES and ASTER and higher than STRATIS (2.9% vs 4.4% in HERMES, 1.4% in STRATIS, 5.3% in ASTER aspiration). The rate of ENT observed in PROMISE was lower than ASTER and higher than STRATIS (1.5% vs 0.8% in STRATIS, 3.7% in ASTER aspiration). It should be noted that ASTER used almost exclusively ACE64 and 5MAX catheters over ACE68 (65 ACE64, 1 ACE68, 63 5MAX devices were used frontline in the aspiration group).
The improvement in revascularization rates observed in PROMISE might be due to the introduction of the ACE68/64 catheters, along with improved technical skills due to the higher volume of endovascular treatment in recent years. On the other hand, improvements of in-hospital processing times implemented after the introduction of new stroke treatment guidelines8–10 as seen in the rapid symptom onset to arterial puncture time (comparison with MR CLEAN, STRATIS, and ASTER in figure 1) might also be correlated with better patient outcomes observed in PROMISE. These observations suggest that the ADAPT technique is an effective method, and that the rate of complications, especially of sICH, is low. This favorable safety profile may arise from the use of the recently introduced large-bore catheters with improved trackability, resulting in less manipulation with multiple devices. Though not a prespecified analysis, no significant differences in safety or effectiveness outcomes were noted between the two catheter groups, ACE64 or ACE68. There were no differences in baseline patient characteristics, except for more cardiac heart failure, less peripheral artery disease, and higher pre-stroke mRS >0, in the ACE68 group. The first-pass success rate with ADAPT was 60.2%, with no significant difference between catheter sizes (58.2% vs 61.2%, ACE64 vs ACE68). Outcomes should be evaluated with caution due to the unknown impact of the baseline covariates. The potential impact of catheter size remains to be elucidated in a larger study with a matched patient population. In the PROMISE study, balloon guide catheters were not used due to lesional aspiration at the proximal face of the clot.
The primary limitation of the PROMISE study was inherent in its study design and lack of a randomized controlled comparison, and the absence of blinding of the 3- month mRS assessment. Although PROMISE study criteria excluded patients with tandem lesions requiring treatment prior to aspiration, nine patients (4.4%) with proximal stenosis were enrolled and five required intervention (two stenting and angioplasty, one stenting, and two angioplasty alone), which may be a lower proportion compared with other studies. The major strengths of this study include evaluation by an independent Core Laboratory and a Clinical Events Committee, prospective and consecutive enrollment of patients to minimize potential bias associated with a registry, all data being monitored during on-site visits, and data collected from real-world clinical practice across Europe.
Summary
As technical and clinical developments in stroke treatment evolve, these results suggest that in current practice, the ADAPT technique with ACE68/ACE64 offers equal or better results compared with other techniques. The findings from the PROMISE study support the use of aspiration with ACE68/ACE64 reperfusion catheters as a frontline therapy in the treatment of patients with ischemic stroke from large vessel occlusion.
References
Footnotes
Contributors All authors made a substantial, direct, and intellectual contribution to the work.
Funding This study was funded by Penumbra, Inc (9508).
Competing interests The following investigators report financial conflicts with this study: PS reports being a consultant for Penumbra and Stryker, and receiving research support from Penumbra, Philips, and Siemens. RP reports proctoring and consulting for Medtronic, Penumbra, and Implemed. WW reports a proctor agreement with Microvention/Terumo, Medtronic, and Phenox, and being a consultant with Penumbra (speaker and chairman fees). JF reports being a consultant for Penumbra, Acandis, Boehringer Ingelheim, Cerenovus, Covidien, Medtronic, Microvention, Route92, and Stryker, and receiving research support from the German Ministry of Science and Education (BMBF), German Ministry of Economy and Innovation (BMWi), German Research Foundation (DFG), European Union (EU), Hamburgische Investitions- und Förderbank (IFB), Medtronic, Microvention, Philips, and Stryker. PM reports speaker and consulting fees from Medtronic, used for stroke education. TK and VMP received funding from the sponsor for imaging core lab oratory activities. VMP reports being a consultant for Penumbra (PROMISE study), Medtronic, Stryker, and Neurovasc. LP received funding from Penumbra for CEC activities and is a consultant for Balt, Cerenovus/Neuravi, and Microvention.
Patient consent Not required.
Ethics approval The local Ethics Committees approved the study, and the patients or their representatives provided written informed consent, according to local regulations.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Because of the sensitive nature of the data collected for this study, requests to access the dataset from qualified researchers trained in human subject confidentiality protocols may be sent to Penumbra, Inc. at promise@penumbrainc.com.