Article Text

Abstract
Background The mechanism of development of delayed cerebral ischemia (DCI) after aneurysmal subarachnoid hemorrhage (SAH) is poorly understood. Inflammatory processes are implicated in the development of ischemic stroke and may also predispose to the development of DCI following SAH. The objective of this study was to test whether concentrations of circulating inflammatory markers (C-reactive protein (CRP), interleukin-6 (IL-6) and interleukin 1 receptor antagonist (IL-1Ra)) were predictive for DCI following SAH. Secondary analyses considered white cell count (WCC) and erythrocyte sedimentation rate (ESR).
Methods This was a single-center case-control study nested within a prospective cohort. Plasma inflammatory markers were measured in patients up to 15 days after SAH (initial, peak, average, final and rate of change to final). Cases were defined as those developing DCI. Inflammatory markers were compared between cases and randomly selected matched controls.
Results Among the 179 participants there were 46 cases of DCI (26%). In primary analyses the rate of change of IL-6 was associated with DCI (OR 2.3 (95% CI 1.1 to 5.0); p=0.03). The final value and rate of change of WCC were associated with DCI (OR 1.2 (95% CI 1.0 to 1.3) and OR 1.3 (95% CI 1.0 to 1.6), respectively). High values of ESR were associated with DCI (OR 2.4 (95% CI 1.3 to 4.6) initial; OR 2.3 (95% CI 1.3 to 4.2) average; OR 2.1 (95% CI 1.1 to 3.9) peak; and OR 2.0 (95% CI 1.2 to 3.3) final value).
Conclusions Leucocytosis and change in IL-6 prior to DCI reflect impending cerebral ischemia. The time-independent association of ESR with DCI after SAH may identify this as a risk factor. These data suggest that systemic inflammatory mechanisms may increase the susceptibility to the development of DCI after SAH.
- Subarachnoid
- Hemorrhage
- Aneurysm
- Inflammation
- Inflammatory Response
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Vasospasm is a phenomenon associated with aneurysmal subarachnoid hemorrhage (SAH), the occurrence of which frequently, but not invariably, leads to the development of delayed cerebral ischemia (DCI). Identifying which patients are at risk of developing DCI after SAH is crucial in developing therapies that can treat, if not prevent, this potentially devastating condition.
Despite major advances in the neurosurgical and endovascular management of intracranial aneurysms, there has been little change in the management of DCI over the last 25 years, apart from the introduction of routine administration of the calcium channel blocker nimodipine.
A number of factors including free radical-mediated injury, microvascular changes, endothelial damage, neurotoxicity from breakdown products of blood, autroregulatory changes, coagulation disturbance and inflammatory processes1 ,2 in combination with vasospasm are all likely to contribute to the subsequent development of DCI following SAH.
Inflammatory processes are implicated in the development of ischemic stroke and levels of plasma inflammatory markers correlate with poor outcome.3 ,4 Inflammation is a key feature of many of the risk factors for ischemic stroke (including atherosclerosis), and it is possible that it acts as a trigger for ischemia. It is feasible that inflammation may increase susceptibility to the development of DCI after SAH. Studying any link between plasma inflammatory markers and the development of subsequent ischemia in the general population is impractical outside very large epidemiological studies. However, patients with SAH who are at a high risk of developing cerebral ischemia within 21 days after ictus provide a unique platform to study the association between levels of peripheral inflammatory markers and the subsequent development of ischemia.
Cerebrospinal fluid (CSF) concentrations of interleukin 6 (IL-6) have been found to be raised following SAH, and appear to be higher in those who later develop evidence of cerebral vasospasm as defined by transcranial Doppler criteria or angiography5–8 or with clinical evidence of DCI.8–12 CSF concentrations of interleukin 1 receptor antagonist (IL-1Ra) are increased following DCI, and elevated concentrations of IL-1Ra have been associated with a poor outcome.13 The source of CSF within these studies varies (and is frequently not stated), and in those studies obtaining CSF directly from the ventricular system a disproportionately high number of patients with poor clinical grade are included.8
Levels of inflammatory markers in the CSF are presumed to reflect inflammatory markers in the brain, although this may not necessarily be the case.14 There is good evidence that systemic inflammation influences inflammatory events in the CNS,15 but the relationship between levels of inflammatory markers in the periphery and in the brain remains unclear.
The few studies of plasma markers in SAH have shown inconsistent results. In one study IL-6 was not increased in peripheral plasma,16 although internal jugular vein concentrations were raised after SAH compared with controls. In other studies, increased concentrations of plasma cytokines were observed after SAH but were not associated with DCI12 or poor outcome.5 ,7 Leucocytosis and elevated C-reactive protein (CRP) may predict a poor outcome following SAH.17–19
In view of the evidence for increased inflammation in SAH and the apparent association with vasospasm, we examined whether systemic inflammation, as identified by increased plasma inflammatory markers, helps to predict which patients with SAH would subsequently develop DCI. It was hoped this might also provide some insights into the nature of the relationship between inflammation and ischemia.
Methods
This was a single-center case-control study nested within a prospective cohort of patients admitted to the Greater Manchester Neuroscience Center at Salford Royal NHS Foundation Trust.
Inclusion criteria and data collection
Adult patients (aged >18 years) with a confirmed diagnosis of SAH (CT imaging and/or positive xanthochromia) and no known concurrent clinical condition likely to interfere with participation or completion of the study within 7 days of ictus were identified between January 2004 and June 2006. Eligibility was restricted to those with an underlying aneurysm who underwent formal angiography.
Clinical details included presenting features, modified Rankin score, World Federation of Neurosurgical Societies grading scale, Fisher grade, aneurysm size and location, detailed demographics, infection and medication screen, atherosclerotic risk factors and physiological parameters.
Patients were admitted to our center at variable times following ictus. Since most DCI events occur before 15 days following SAH, and we were interested in whether inflammation predicted this event, initially blood was taken daily up to 13 days from ictus. Because of the variable admission time and concern that angiography could itself provoke an inflammatory response, it was planned to examine the temporal profile of inflammatory markers following recruitment of the first 40 patients. Following this, blood was drawn from the rest of the cohort prior to angiography, which represented an easily identifiable and practical time point in that all participants were under observation from this time. Further blood samples were taken 1 day after angiography, then at days 3, 5, 7, 9, 11 and 13 after angiography or until day 13 after ictus if this was sooner.
Daily clinical assessments were performed up to 15 days after ictus to assess the presence of DCI. DCI was defined as one or more of a new focal neurological deficit, a 2-point drop in the Glasgow Coma Scale or a new infarct on brain imaging not visible on the admission or post-intervention scan in the absence of seizures, metabolic disturbance, infection or hypoxia. A CT scan was performed in all cases to exclude a further hemorrhage or hydrocephalus. Any event within 24 h of coiling or clipping was excluded to avoid including ischemic events due to the procedure itself rather than DCI. All presumed events were detailed in an anonymized event report form and assessed by the chief investigator and a consultant neurovascular surgeon independent of the study. If agreement between the two neurosurgeons could not be reached, the patient was excluded from further analysis. At 6 months after ictus the extended Glasgow Outcome Score was measured.
Case-control selection
Cases were defined as those who developed DCI during the 15 days following ictus. Controls were selected randomly from participants who had undergone the same blood sampling protocol, were still under observation at the time of the case DCI and had definitely not yet had DCI. Patients were excluded as possible controls if they had a significant inflammatory event (eg, myocardial infarction or severe infective episode leading to clinical deterioration) or the absence of DCI could not be confirmed. Up to double the number of controls were selected for each stratum of cases defined by the day after angiography on which the DCI occurred. To avoid bias, incidence density sampling was used in which all eligible participants were potential controls for each stratum even if selected previously as controls for earlier strata or contributing themselves as cases in subsequent strata.
Laboratory analyses
Venous blood was collected into ethylenediamine tetra-acetic acid (EDTA) for full blood count analysis (Coulter Gen-S Analyser; Coulter, California, USA), tri-sodium citrate for determination of erythrocyte sedimentation rate (ESR) (Westergren method) and serum gel tubes for the biochemical profile (Roche Integra 700 analyser; Roche, UK).
A further 10 ml of blood was collected into 100 µl pyrogen-free heparin, cooled to 4°C and centrifuged for 15 min at 2000 g. Plasma was decanted and stored at −70°C prior to analysis of inflammatory markers. Measurement to determine the profile of IL-6 in the first 40 subjects was undertaken essentially as previously described.20 Subsequently, the assay was modified to use PeliPair anti-IL-6 antibodies (M9316, Sanquin, Amsterdam) and development with Zymed steptavidin-horseradish peroxidise conjugate (ZyMak grade, Zymed Laboratories, San Francisco, USA). Measurement of CRP was by competitive ELISA, as described previously.21 Measurement of IL-1Ra was as described previously.22 After allowing for sample dilution, minimum assay sensitivities were 0.7 pg/ml for IL-6, 366 µg/l for CRP and 31 mg/l for IL-1Ra. Interassay coefficients of variation, determined in the appropriate working range, were 12% at 4 pg/ml to 6% at 111 pg/ml for IL-6; 16% at 511 µg/l to 15% at 29.05 mg/l for CRP; and 16% at 139 pg/ml to 7% at 1045 pg/ml for IL-1Ra. Laboratory standards for IL-6, IL-1Ra and CRP were calibrated against WHO international standards for 89/548, 92/644 and 85/506, respectively, from the National Institute for Biological Standards Control (South Mimms, UK). Fibrinogen was measured as previously described.23
Statistical analysis
For each case and selected control, summary statistics were calculated based on their inflammatory marker profiles until the time of DCI. Logarithms of concentrations were calculated prior to calculation of summary statistics to normalize their distributions. Peak, average, initial, final and final rate of change (average increase per day over last two readings) were calculated for each profile. The association of each summary statistic with DCI was assessed separately in multifactorial conditional logistic regression, adjusting for age and sex. The use of conditional logistic regression was required to account for the matching of controls to cases within each stratum defined by time to DCI following angiography. The associations were expressed as OR with 95% CIs and p values. Three inflammatory markers were deemed ‘primary’ to limit the possibility of spurious statistical significance arising by chance, given the large number of potential analyses. Interpretation of results for ‘secondary’ markers is more cautious for this reason.
Results
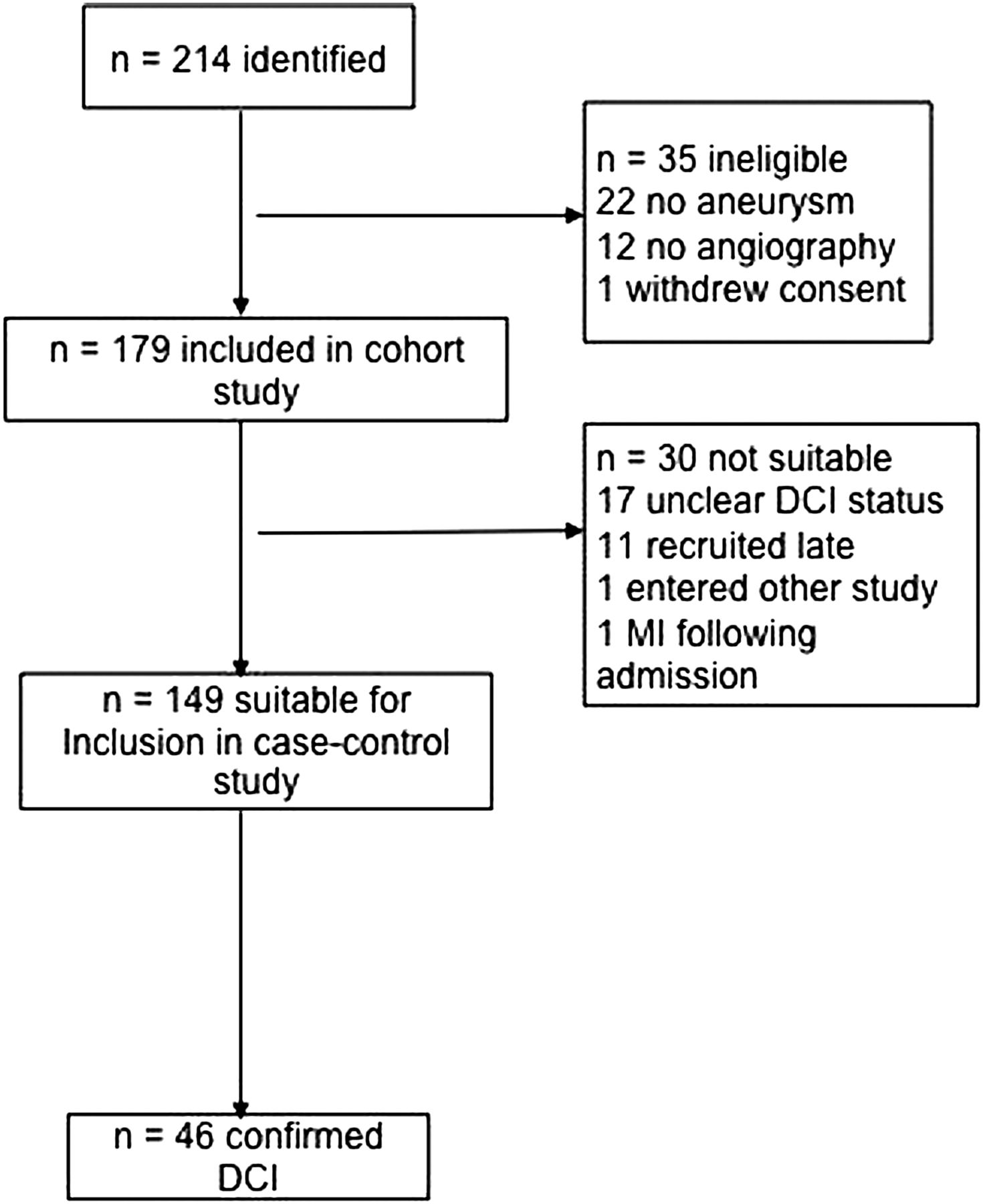
We identified 214 possible participants. Of these, 35 were ineligible or withdrew consent, leaving 179 recruited to the study. A further 30 could not be included in the analyses (figure 1), leaving 149 patients eligible for analysis of whom 46 (31%) developed DCI 3–13 days after SAH (mean time to onset 8 days). The characteristics of the study population (n=149) are given in table 1.
Patient characteristics

Flow chart of selection of subgroups and reason for exclusion. DCI, delayed cerebral ischemia.
A review of the inflammatory marker profile in the first 40 patients showed that a rise in inflammatory markers generally occurred following angiography (figure 2). Therefore, as DCI was the event of interest and ictus was not critical to the analysis of whether inflammatory markers were predictive of DCI, we used time of angiography as the common baseline for all patients.

Temporal profiles of C-reactive protein (CRP), interleukin 6 (IL-6) and interleukin 1 receptor antagonist (IL-1Ra) after subarachnoid hemorrhage plotted to day of angiography for first 40 patients and box and whisker plots of data for all delayed cerebral ischemia cases and their matched controls.
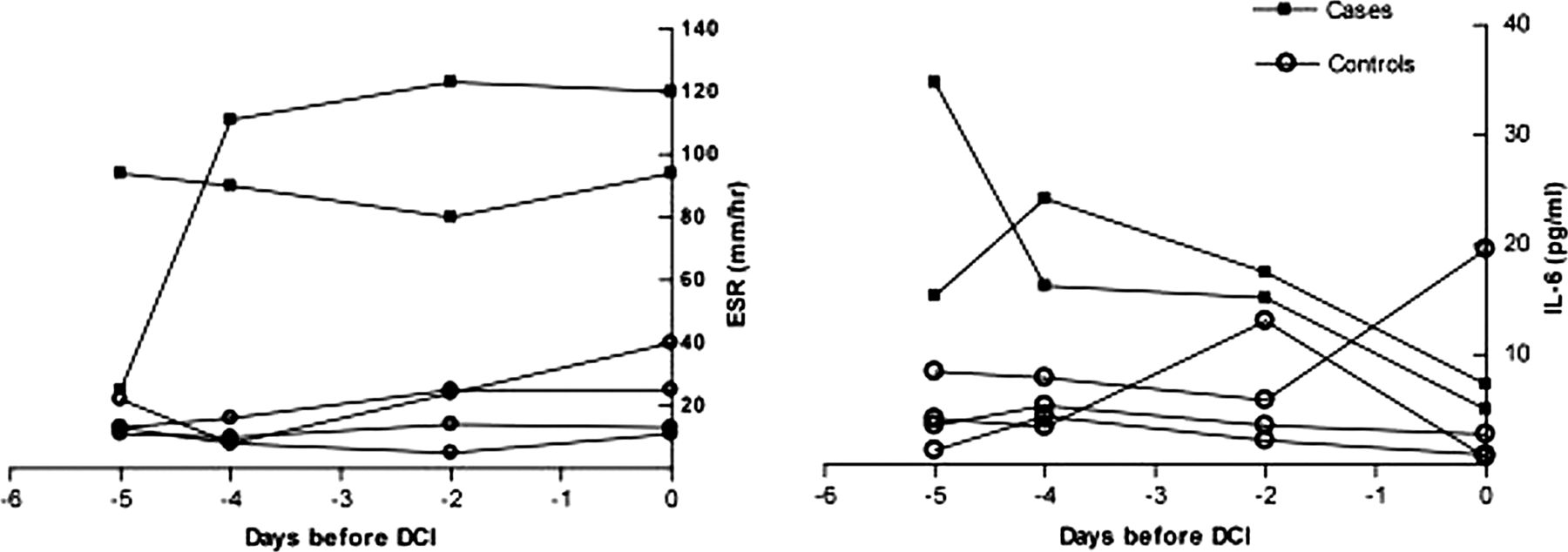
Of the 15 summary statistics for the three primary markers examined (IL-6, IL-1Ra and CRP), only the final rate of change of IL-6 was significantly associated with DCI (table 2) with IL-6 falling less steeply in those about to experience DCI. In secondary analyses, final and final rate of change of white cell count (WCC) were found to be associated with DCI. A high ESR throughout was also consistently associated with DCI. Figure 3 shows an example of a ‘case-control strata’ (two cases and four controls) for IL-6 and ESR. We examined whether the fibrinogen concentration might be responsible for the relationship with ESR, but there was no significant association between initial fibrinogen concentration and DCI and the correlation between fibrinogen and ESR was weak.
Results of case-control analyses for inflammatory markers
{kind=link}
{kind=link}
{kind=link}
Example profiles of case-control strata for erythrocyte sedimentation rate (ESR) and interleukin 6 (IL-6). DCI, delayed cerebral ischemia.
Discussion
Most clinical studies examining peripheral inflammatory markers and cerebral ischemia have been performed on patients who have had an acute ischemic stroke in the period after the acute stroke has occurred. Elevation of peripheral IL-6,24–26 IL-1Ra24 ,27 and CRP20 have been reported after ischemic stroke and are related to clinical outcome. The ESR and WCC were also independently associated with infarct volume and functional outcome after stroke when examined retrospectively,28 and elevated ESR has been shown to predict the short-term outcome following stroke in a large prospective study (n=208) when measured within 72 h of symptom onset.29 Additionally, ESR was identified in the Reykjavik study as an independent predictor of both ischemic heart disease and fatal stroke in a follow-up period of >20 years.30
Although ESR and CRP have been shown to have some predictive value for ischemic events,30 ,31 it is difficult to gather data in the immediate period preceding a stroke. In contrast, patients with SAH offer the opportunity to evaluate the inflammatory response preceding the development of an ischemic event in a relatively young previously asymptomatic group.
An association between CSF IL-6 levels after SAH with symptomatic vasospasm,5 DCI12 or outcome5 ,7 ,8 has been described. A recent paper reported that IL-6 levels in the CSF on day 3 after SAH may predict the development of vasospasm as defined by transcranial Doppler examination.32
CSF concentrations of IL-1Ra on days 4–10 have been reported to be associated with a poor outcome after SAH.13 We failed to demonstrate any predictive association between plasma IL-1Ra concentrations after SAH and the development of DCI.
Of the primary outcome measures, only the final rate of change of IL-6 showed a significant association with DCI (OR 2.3, 95% CI 1.1 to 5, p=0.03).
The IL-6 concentration is elevated after SAH compared with healthy controls. Levels of IL-6 are then seen to decline over 7–10 days after the SAH.33 It may be that a new (possibly smaller) signal from impending DCI has the effect of arresting this decline. The greater gradient in IL-6 in cases demonstrated in this study may therefore be related to an acute inflammatory event associated with the subsequent development of ischemic damage. Some recent experimental data indicated that neutralization of IL-6 could be effective in reducing cerebral ischemia after middle cerebral artery occlusion.34
There is an extensive body of literature identifying CRP as a predictor of cardiac events31 ,35 and as an independent predictor of outcome after stroke.36 ,37 However, the present study failed to identify any predictive value of CRP following SAH with the risk of DCI. This suggests that the size of an inflammatory response following initial hemorrhage does not influence whether DCI will follow, although a statistically significant association between DCI and CRP concentrations on days 5, 6, 7 and 8 in 88 patients following SAH has been reported.19 In this study, a similar association between DCI and WCC on days 1, 4, 5, 6 and 7 was observed. However, these patients all underwent surgery rather than endovascular coiling, and both CRP and WCC measurements were included for those patients who had already developed DCI.
The rate of change of WCC and final WCC were found to be significantly associated with the development of DCI, as shown in table 2. This is supported by data from other studies where no predictive value was seen in admission WCC for patients with SAH who went on to develop DCI but a significant rise in WCC was found at the time of ischemia.18 ,38 It is also in accord with the changes in circulating IL-6 that we report, which may suggest that a change in inflammatory markers reflect events occurring just before the development of the ischemic deficit.
ESR was the inflammatory marker most consistently associated with the development of DCI. Initial, average, peak and final ESR were all significantly associated with the subsequent development of DCI. Rate of change was not associated with the development of DCI, presumably because of the pre-existing elevation of the marker in response to the initial hemorrhage and because it changes more slowly in response to inflammation. We are unaware of any other studies that have shown an association between the risk of DCI and ESR, although we found a consistent result in a study examining the risk of a vascular event following a transient ischemic attack.23
It is recognized that elevation of ESR may be due to elevation of plasma fibrinogen,39 which could favor the proposal that coagulopathy after SAH contributes to DCI.40 The role for increased coagulation, in combination with inflammation, has recently been reviewed as a potential explanation for DCI.41 Fibrinogen, in conjunction with D-dimers, has been found to be higher in patients with SAH who develop DCI.42 D-dimers, and hence activation of fibrinolysis, are associated with severe delayed ischemic deficit, clinical outcome, patient fatality and the development of cerebral infarction after SAH.43 ,44 To address the possibility that fibrinogen was responsible for increased ESR, fibrinogen assays were performed in all case-control strata within the study population. No significant association was seen between initial fibrinogen levels and the development of DCI, and there was little correlation between ESR and fibrinogen concentrations. This may suggest that the relationship between DCI and ESR was independent of fibrinogen; however, samples were available only from the initial time point and the standard functional assay had to be adapted to use EDTA-anticoagulated blood. It is noteworthy that 79% (118 patients) of the study population were current or ex-smokers, so ESR may reflect pre-existing vascular disease. However, after adjustment for smoking status, the associations of ESR (initial value, final value, peak and average) remained significant.
Conclusions
Systemic inflammation may increase susceptibility to the development of DCI after SAH. The change in IL-6 and WCC immediately before DCI may identify a proximal inflammatory event associated with the ischemic episode. In contrast, the time-independent relationship of ESR with DCI after SAH may identify this as a risk factor for DCI that is not simply related to other measures of the post-SAH inflammatory response. Although the results should be interpreted with caution because of the multiple comparisons and associations with secondary measures, they provide support for the concept that measures to reduce inflammation should be evaluated as a protective therapy in this devastating condition.
Acknowledgments
We acknowledge the support of the University of Manchester for making this research possible and the neurosurgical patients and staff at Salford Royal Hospital.
References
Footnotes
-
Contributors CJM, PJT, SH, AV, ATK and NJR designed the study and were involved in its implementation. CJM, SC and DS were involved in recruitment of patients to the study, data and clinical sample collection. KJI and SH were involved in sample processing and analysis. CJM and AV were involved with statistical analysis of results. The draft paper was written by CJM with input from SH, ATK, PJT and NJR.
-
Funding This work was supported by a grant awarded by the Sir Jules Thorn Charitable Trust, Thorn Trust Ref No 02/JTA. University research account number: R010453 (applicants: NJR, PJT, SH, AV).
-
Competing interests None.
-
Ethics approval Ethical approval was obtained from the Bolton Research Ethics Committee via the National Research Ethics service and University of Manchester Research Ethics Committee.
-
Informed consent Obtained from patients or their representative.
-
Provenance and peer review Not commissioned; externally peer reviewed.