Article Text

Abstract
Background The use of flow diversion to treat intracranial aneurysms has increased in recent years.
Objective To assess the safety and angiographic efficacy of the p64 flow modulation device.
Methods Diversion-p64 is an international, prospective, multicenter, single-arm, study conducted at 26 centers. The p64 flow modulation device was used to treat anterior circulation aneurysms between December 2015 and January 2019. The primary safety endpoint was the incidence of major stroke or neurologic death at 3–6 months, with the primary efficacy endpoint being complete aneurysm occlusion (Raymond-Roy Occlusion Classification 1) on follow-up angiography.
Results A total of 420 patients met the eligibility criteria and underwent treatment with the p64 flow modulation device (mean age 55±12.0 years, 86.2% female). Mean aneurysm dome width was 6.99±5.28 mm and neck width 4.47±2.28 mm. Mean number of devices implanted per patient was 1.06±0.47, with adjunctive coiling performed in 14.0% of the cases. At the second angiographic follow-up (mean 375±73 days), available for 343 patients (81.7%), complete aneurysm occlusion was seen in 287 (83.7%) patients. Safety data were available for 413 patients (98.3%) at the first follow-up (mean 145±43 days) with a composite morbidity/mortality rate of 2.42% (n=10).
Conclusions Diversion-p64 is the largest prospective study using the p64 flow modulation device. The results of this study demonstrate that the device has a high efficacy and carries a low rate of mortality and permanent morbidity.
- aneurysm
- flow diverter
- stent
Data availability statement
Data are available upon reasonable request. Data will be available after a resonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The development and introduction of flow diverter stents into clinical practice was a pivotal moment in the treatment of cerebral aneurysms.1 2
It is now understood that devices constructed with an increased number of wires have a greater flow diverting effect3 and a lower variance in the flow diverting effect when there are changes in the caliber of the underlying vessel or in curved vessels.4
The p64 flow modulation device (p64 FMD, phenox GmbH, Germany) is constructed from 64 braided nickel-titanium wires and designed to provide optimized flow diverting properties. The device is approved for the treatment of intracranial aneurysms and dissections in Europe, and a growing body of literature supports its long-term efficacy.5–12 Diversion-p64 was a single-arm, international, multicenter, prospective study to assess the safety and efficacy of the p64 FMD and is the largest prospective study on flow diversion to date.
Methods
Study design, enrollment, and patient selection
Diversion-p64 (ClinicalTrials.gov identification code: NCT02600364) was a single-arm, international, multicenter, prospective study to assess the safety and efficacy of the p64 FMD. Between December 2015 and January 2019 patients were enrolled at 26 centers from 10 different countries, including eight European countries, Argentina and Russia (online supplemental figure 3). Recruiting sites had to be familiar with the p64 FMD. All participating sites were required to perform five ‘lead-in cases’ with p64 FMD under the supervision of an experienced proctor prior to study participation.
Supplemental material
The eligibility criteria are listed in the online supplemental material. The per-protocol population contains all enrolled patients who met all eligibility criteria and had at least one p64 FMD implanted at the end of the intervention.
Study device
The p64 FMD received the Conformité Européenne (CE) mark in 2012. The p64 FMD is constructed from 64 braided nickel-titanium wires and is available in diameters ranging from 2.5 to 5 mm and lengths ranging between 9–36 mm. Proximally, the 64 wires form eight bundles each of which has a proximal radiopaque marker. Two platinum wires allow radiopacity and visibility of the device in vivo. The p64 FMD is the only flow diverter available on the market that is mechanically detachable and this unique feature allows the device to be completely unsheathed, resheathed and to be delivered through a 0.027” inner diameter microcatheter.
Baseline assessments
Before placement of the p64 FMD, patients underwent a neurological assessment using the modified Rankin Scale (mRS) and National Institutes Health Stroke Scale (NIHSS). Medical history and concomitant medications were recorded. In case of ruptured aneurysms, the World Federation of Neurosurgeons classification was also recorded.
Dual antiplatelet therapy
Consistent with the instructions for use for the p64 FMD, dual antiplatelet therapy was advised. The use of heparinization during the procedure was encouraged and the choice and duration of dual antiplatelet therapy was according to the local site-specific standard operating procedures. Preoperative testing of responsiveness to antiplatelets with point-of-care testing equipment. such as VerifyNow (Accumetrics, San Diego, California, USA), was encouraged but not mandatory.
Follow-up assessments
Patients were assessed prior to discharge and when new neurological deficits were seen, cross-sectional imaging of the brain, with either CT or MRI, was performed. Two clinical and imaging follow-ups were scheduled between 3 and 6 months and 7 and 12 months according to site-specific standard operating procedures.
Safety reporting
Investigators were required to report all adverse events and serious adverse events. An independent Clinical Events Committee (CEC) adjudicated all relevant adverse events and serious adverse events reported by the investigators or by the core laboratory and determined whether these were related to the device, the procedure, or both. The CEC consisted of three independent physicians with specialist expertise in neurological diseases and vascular neurology (composition listed in online supplemental material). The core laboratory reviewed the image material of all procedures, and any complication depicted on imaging that was not yet known from event reporting by the investigators was additionally presented to the CEC for adjudication.
Study endpoints
Primary efficacy endpoint
The primary efficacy endpoint was the complete occlusion of the intracranial aneurysm, based on the Raymond-Roy Occlusion Classification.13 An independent core laboratory, blinded to all clinical information, reconfirmed the aneurysm type and location, matched the eligibility criteria, and assessed the aneurysm at both follow-up visits.
Before the core laboratory readers started their review, they were trained on the study protocol, schedule of imaging assessments, and aligned on definitions and reading from a general perspective. All core laboratory members are licensed and practicing interventional neuroradiologists, each with over 10 years of experience in interventional neuroradiology and experience as core laboratory reader in previous studies. The members of the core laboratory were not practicing at any of the investigational sites (composition listed in online supplemental material). Two core laboratory readers performed a review of all patients independently from each other. The resulting data were compared, and in cases of discrepancy a third reader judged the case as an adjudicator.
Primary safety endpoint
The primary safety endpoint was the incidence of major stroke (ischemic or haemorrhagic) related to treatment of the target aneurysm, defined as an increase in the NIHSS score by 4 points or neurologic death at 3–6 months, with a secondary safety endpoint at 7–12 months after treatment.
Secondary endpoints
Secondary efficacy endpoints included technical feasibility to deliver and detach the p64 FMD at the correct location. Other secondary safety endpoints included evidence of aneurysm recanalization or regrowth, intraprocedural complications such as vessel or aneurysm perforation, and thromboembolic complications as well as delayed postprocedural complications—for example, delayed aneurysmal rupture and infarction detected on follow-up imaging.
Statistical analysis
Demographics, baseline data, and procedural characteristics were summarized and reported as mean±SD and range for continuous variables. Categorical data were summarized using numbers and percentages.
The main hypothesis of the study was that the rate of neurological death and major stroke within 3–6 months after treatment with the p64 FMD in patients with aneurysms in the anterior circulation was not inferior to the weighted mean rate of neurological death and major stroke within 6 months from three relevant studies at the time of study design and recruitment. The weighted average rate of neurological death and major stroke within 3–6 months after treatment was calculated as 6.6% and derived from the results of three key publications using the Pipeline embolization devices (PED, Medtronic): Pipeline for the Intracranial Treatment of Aneurysms (PITA), Pipeline for Uncoilable or Failed Aneurysms (PUFS), International Retrospective Study of the Pipeline Embolization Device (IntrePED).14–16 A clinically acceptable non-inferiority margin of 4% was used. Based on this, a sample size of 348 patients completing the 3–6 months' follow-up was calculated to provide a power of 80% to meet the primary safety endpoint.
To evaluate non-inferiority a one-sided, one-sample binomial test was performed. The statistical analysis was performed in RStudio (version 4.0.3).
Results
A total of 420 patients met all eligibility criteria and underwent treatment with the p64 FMD and constitute the per-protocol group on which this analysis has been performed.
The mean age of patients was 55±12.0 years (range 24–88) and 362 (86.2%) were female. The mean aneurysm dome width was 6.99±5.28 mm (range 1.0–32.0 mm), with mean neck width 4.47±2.28 mm (range 1.0–15.0 mm). Very small aneurysms (<4 mm) accounted for 31.4% of aneurysms. Of those 131 small aneurysms, 22 (16.8%) were previously treated (21 coiled and one clipped) and 17 (13%) were ruptured (one acutely and 16 previously). The majority of saccular aneurysms (64%) were small (<7 mm) and 61.1% of aneurysm had a wide-neck (dome to neck ratio <1.5). Blister-like and fusiform aneurysms represented 3.8% and 1.2%, respectively, while dissecting aneurysms and segmental diseases were 1.0% each. The most common aneurysm locations were paraophthalmic (59.3%) and posterior communicating artery (16.9%). Most of aneurysms were asymptomatic (77.4%) and found incidentally (66.4%). The vast majority of aneurysms were unruptured (93.3%) with seven aneurysms (1.67%) treated within 21 days of rupture. Most aneurysms had not been previously treated (86.9%). Of the previously treated aneurysms, the majority were coiled (n=52/55, 94.5%). Baseline aneurysm characteristics are summarized in table 1.
Baseline aneurysm data
A total of 445 p64 FMDs were implanted and in the vast majority, a single p64 FMD was implanted (96.4%), with 1.06±0.47 used for each patient. Adjunctive coiling was performed in 59/420 (14.0%) cases. In 98.1% of cases, the p64 FMD was deployed at the desired location, with 97.4% with correct opening of the implanted p64 FMD (online supplemental table 1). In total, 442 implanted devices were detached correctly (99.3%).
Intraprocedural complications
In total, the core laboratory assessment determined that 23 patients had an intraprocedural complication, with the most common being thromboembolism (n=17/420, 4.0%). These were managed using a variety of different strategies depending on local practice but included the use of antiplatelet agents, anticoagulants, and balloon angioplasty. Intraoperative vessel perforation occurred in two patients and intraoperative aneurysm perforation in one patient. Intraoperative side branch occlusion occurred in two patients and difficulty of device detachment was recorded in three cases.
Primary effectiveness endpoint
Angiographic follow-up data were available for 357 patients (85.0%) at the first follow-up performed at mean 145±43 days (range 27–214 days). At this point, complete occlusion and residual neck were reported in 71.7% (n=256) and 4.5% (n=16) of cases, respectively, leading to 76.2% (n=272) cases of adequate occlusion.
At the second follow-up, performed at mean 375±73 days (range 245–579 days), angiographic data were available for 343 patients (81.7%). The reasons for the missing angiographic assessments are reported in online supplemental figure 1. Complete aneurysm occlusion (figure 1) and residual neck were seen in 83.7% (n=287) and 2.3% (n=8) of cases, respectively, leading to 86.0% (n=295) of adequate occlusion.
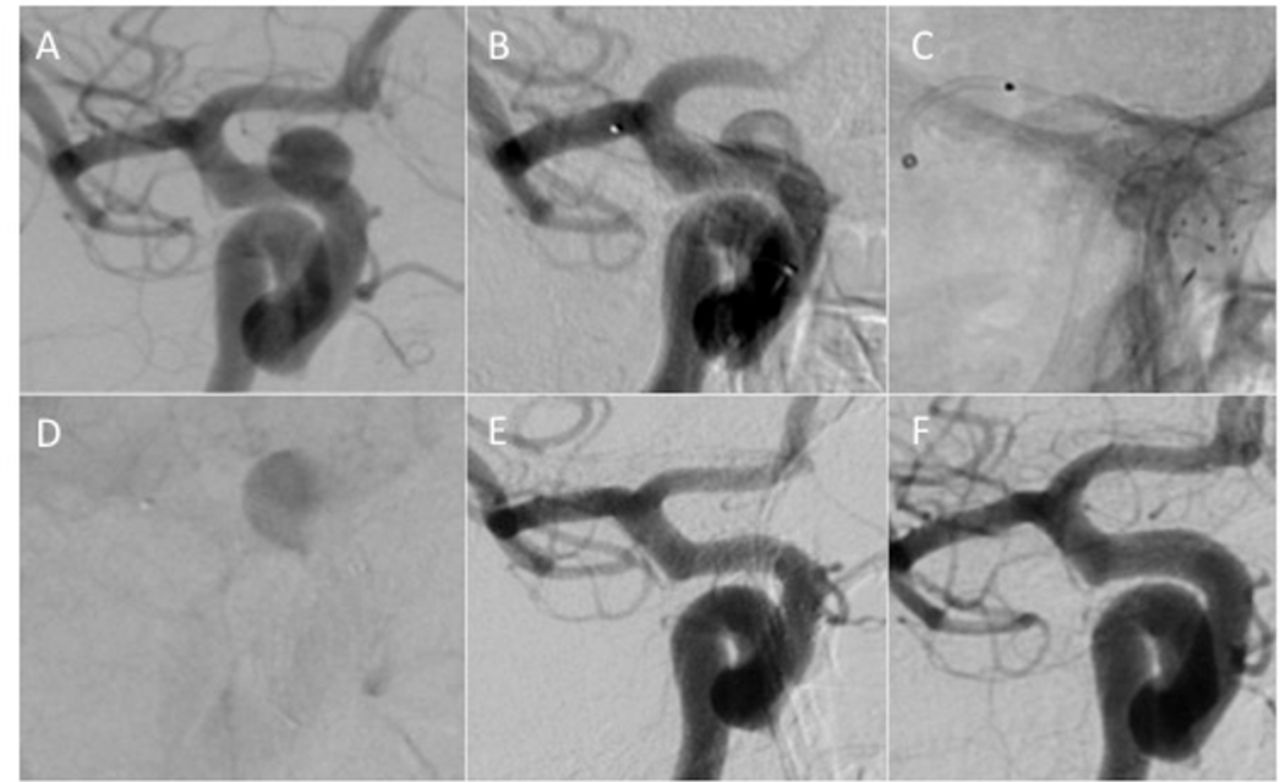
{kind=link}
A 6 mm right-sided, symptomatic paraophthalmic, saccular, aneurysm (A). The aneurysm was treated using a single 4×12 p64 flow modulation device. A satisfactory position of the device (B) was confirmed on unsubtracted images (C). Significant contrast stagnation (D) could be seen at the end of the procedure. Control angiography performed at 4 months postprocedure showed complete occlusion of the aneurysm (E), that was confirmed on delayed angiography performed 10 months post-rocedure (F).
Primary safety endpoint
Online supplemental figure 2 shows the disposition of safety data collected at three different time points during the study. Safety data were available for 413 patients (98.3%) at the first follow-up postprocedure. Of these 413 patients a major procedure-related stroke occurred in eight patients (1.9%). All were thromboembolic complications; seven occurred intraprocedurally or periprocedurally, resulting in infarction with mass effect in three cases, with subsequent parenchymal hemorrhage in one case. One of the strokes occurred secondary to non-compliance with medication. Two patients were discharged with mild (NIHSS score 2) or no symptoms (NIHSS score 0) of stroke.
Mortality rate was 0.97% (n=4) related to a major stroke within the territory of the implanted device in two patients. In one case, a type A aortic dissection was discovered in the early postoperative period that resulted in myocardial infarction and death. In the last patient, death occurred 3 weeks postprocedure, secondary to a subdural hematoma that was thought to be caused by antiplatelet medication. The composite morbidity/mortality rate was therefore 2.42% (n=10/413) at the first follow-up. Comparing this rate with its 95% confidence interval (1.17% to 4.43%) to the target of 6.6% adding the non-inferiority margin, results in a significant p value of <0.0001. The 95% exact upper CI was 4.43%, which was below the threshold of 10.6%; therefore, the primary safety endpoint of the study was met.
No further episodes of major stroke or death occurred between the first and second follow-up with data available for 372 patients. Therefore, by the time of the second follow-up the composite endpoint was met in 2.69% of patients, with mortality seen in 1.08% and major neurological morbidity in 2.15%.
Secondary endpoints
No delayed aneurysmal ruptures or parenchymal hematomas were seen. A total of 23 patients with minor strokes at the first follow-up with one further minor stroke by the second follow-up were reported. Therefore, the minor stroke rate was 6.4% (n=24). Ischemic stroke was found in 22 cases (91.7%) within the first 4 days after implantation and in the remaining two cases there was no correlation between the clinical symptoms and the corresponding image material. Of those 22 patients, two received no antiplatelet medication before intervention and no additional bolus of antiplatelet medication during the procedure, five patients were receiving single antiplatelet therapy, and the remaining 15 were receiving dual antiplatelet therapy. In 19 of 24 patients, a P2Y12 receptor inhibition test was performed before implantation, and all were responders. All of those minor strokes and silent infarctions occurred in the same territory of the treated vessel, probably thromboembolic in origin. Of all the patients who had a minor stroke (n=24), five were clinically silent (20.8%) and 12 had transient clinical symptoms that completely resolved (50%). In total, 23 of the 24 patients (95.8%) who had minor strokes were reported to have an mRS score of 0 and one patient an mRS score of 2.
In-stent stenosis
At the initial angiographic follow-up, in-stent stenosis of any degree was seen in 15.4% of cases (n=55/357) and the majority of these cases (n=31/55) were mild (<50%). Any degree of in-stent stenosis was seen in 8.7% of patients at the second follow-up (n=30/343). The majority of cases (5.5%, n=19/343) represented mild stenosis with only a single case of severe stenosis (≥75%).
Aneurysm growth, recanalization, and re-treatments
In total there were two aneurysm growths, one seen at the first follow-up (0.3%) and one seen at the second follow-up (0.3%). There were four aneurysm recanalizations (blood flow into the aneurysm increased compared with previous visit), two at the first follow-up (0.6%) and two at the second follow-up (0.6%).
Eight planned (1.9%) and two unplanned (0.5%) re-treatments were carried out by the first follow-up and a further two planned re-treatments (0.5%) by the second follow-up.
Discussion
Diversion-p64 is the largest prospective study looking at flow diversion using the p64 FMD to date. The results of the study were compared with similar devices available on the US and EU market.
Several previous publications have looked at the p64 FMD, but these reports have been retrospective with no core laboratory, no CEC, and no source data verification.6 7 10–12 Aguilar Pérez et al 9 recently published a retrospective, single-center series of 530 patients harboring 617 unruptured saccular aneurysms (or those treated >30 days post-rupture), located in the anterior circulation and treated with at least one p64 FMD. The majority of the aneurysms (562, 91.1%) were <10 mm, with an average aneurysm dome size of 4.8 mm (range 1–27 mm), and the p64 FMD procedure was the first treatment performed for 515 (83.5%) of the aneurysms. Mid-term and delayed angiographic follow-ups documented a progression of complete occlusion from 76.6% to 86.4%, residual neck remnants in 8.1% and continued dome filling in 5.5% of aneurysms. The overall rate of thromboembolic complications was 4.8%. The permanent morbidity/mortality rate was 2.4%. Prior studies have shown similar results with progressive aneurysm occlusion over time and similar morbidity/mortality rates.6 Overall, the results of Diversion-p64 are similar to these previous studies, with a low permanent morbidity/mortality of 2.4% and adequate occlusion rate of 86% seen on second angiographic follow-up. These results highlight the overall safety and efficacy of the p64 FMD.
The results of the Diversion-p64 study will be compared with similar prospective, multicenter studies using alternative flow diverter stents. A special focus on primary safety and efficacy endpoints is summarized in table 2. Even though those studies recruited mainly small and large wide-neck unruptured aneurysms the data in table 2 should be taken with caution as investigators used different size grading scales, which makes relevant clinical performance comparisons between devices hazardous. For each of them, the narrative will be restricted to the main findings of the study.
Safety and efficacy results of Diversion-p64 in comparison with similar prospective non-randomized studies involving other flow diverters
The PREMIER study17 was a prospective, multicenter, single-arm trial to evaluate the safety and effectiveness of the Pipeline embolization device (PED, Medtronic) in the treatment of aneurysms measuring ≤12 mm with a wide-neck, which enrolled 141 patients. The median aneurysm size was 4.6 mm (mean 5.0±1.92 mm), with the majority of the aneurysms treated being smaller than 7 mm (n=119, 84.4%).
The safety and efficacy analysis (SAFE) of the flow direction endoluminal device (FRED) in the aneurysm SAFE study18 was a prospective, single-arm, multicenter, observational French study that enrolled patients with 103 aneurysms, either unruptured (n=76, 73.8%) or recanalized (n=27, 26.2%). The majority of aneurysms (68.9%) were small (<10 mm) and wide-necked (96.1%). The most common locations were the cavernous and supraclinoid segments (83.5%) of the internal carotid artery.
Taschner et al 19 recently published results from a prospective, international, multicenter trial assessing the Derivo embolization device (DED, Acandis, Pforzheim, Germany). Inclusion criteria allowed enrollment of unruptured aneurysms of any size located within either the anterior or posterior circulation in patients with a mRS score of ≤1. The study enrolled 119 patients with median aneurysm and neck size of 14.2±16.9 mm and 7.7±9.6 mm, respectively, with predominantly saccular morphology (80%) and anterior circulation (88%).
Wakhloo et al 20 published a prospective, international multicenter, single-arm study assessing the Surpass (Stryker Neurovascular, Kalamazoo, USA) device intended for the treatment of intracranial aneurysms of the anterior and posterior (14.5%) circulations. Successful flow-diverter delivery was achieved in 161 patients with 186 aneurysms (98%). The majority of aneurysms were <10 mm (n=117, 62.9%). Clinical follow-up was available for 150 patients (median 6 months; range 1–38 months) and the primary safety endpoint (neurological death or major stroke) was met in 18 patients (11%). Permanent neurologic morbidity and mortality were 6% and 2.7%, respectively. Morbidity occurred in 4% and 7.4% of patients treated for aneurysms of the anterior and posterior circulation, respectively. Of the 186 aneurysms treated, angiographic follow-up data were available for 158 aneurysms (86.8%) at a median of 6 months (range 1–38 months) with 75% complete occlusion (n=118).
In comparison with these previous studies, the results from Diversion-p64 are favorable. The mean aneurysm size in Diversion-p64 was 6.99±5.28 mm, which is comparable to the aforementioned studies. Although a substantial number of patients were lost between the first (6 months; 413/420, 93.3%) and last safety follow-up (12 months; 372/420, 88.6%), only a single minor stroke was recorded within this time frame. Even though primary safety endpoints could not be compared, owing to a lack of a common definition, the cumulative morbidity/mortality is comparable to similar studies (table 2).
Angiographic aneurysm occlusion rates were also very favorable, with 83.7% of aneurysms occluded at the second angiographic follow-up, and 86% achieving adequate occlusion. In comparison with these previous studies, taking into account that Diversion-p64 has recruited the largest number of very small (<4 mm) aneurysms, the complete occlusion rate for the p64 FMD exceeds that of the other devices on the market over a similar time span. The in-stent stenosis rate in Diversion-p64 is also comparable to rates reported for other devices. Ravindran et al 21 recently described an in-stent stenosis rate of 7.4% in a cohort of 155 patients and 8.8% in their systematic review. In the earlier publication of John et al,22 38% of patients had some degree of luminal narrowing post PED implantation with 9.8% having >25% in-stent stenosis. Similar rates of in-stent stenosis have been reported for the FRED (7.6%)23 and the DED (6.6%).24
Limitations
The main limitations of Diversion-p64 are the non-randomized nature and the lack of a control arm with inherent risk of selection bias. This has been acknowledged as several flow diverter stents were available in most of the participating centers and flow diverter selection was left to the treating physician. This bias can always be a component in a non-randomized study with flow diverters and was reduced by the number of 26 participating sites in 10 different countries. There is no long-term follow-up and heterogeneous use of antiplatelet medications and their duration. Angiographic imaging was not available for all patients and this might affect the overall occlusion rate. However, we believe that this is unlikely to be the case given previous studies on flow diversion and the improved occlusion rates seen on longer follow-up in the study. Furthermore, the majority of aneurysms included in the study were <10 mm, although most aneurysms were wide-necked. This was the case in several similar studies on flow diversion and the results of Diversion-p64 may not directly translate for occlusion rate and safety for large and giant aneurysms.
Conclusion
Diversion-p64 is the largest prospective study on the p64 FMD. The results of Diversion-p64 demonstrate one of the lowest rates of morbidity/mortality seen in any prospective study on flow diversion. It can be concluded that the device has a high efficacy and excellent safety profile that is comparable to other devices.
Supplemental material
Data availability statement
Data are available upon reasonable request. Data will be available after a resonable request.
Ethics statements
Patient consent for publication
Ethics approval
The study protocol and the informed consent form were submitted to the local ethics committee for favourable opinion. Each ethics committee approved the study protocol and the informed consent rorm. All patients gave informed consent before taking part in the study. The following ethics committees were involved: Leading ethics committee: State Chamber of Physicians Baden-Wuerttemberg (DE/EKBW01), Ref-No. F-2015-066. Further ethics committees: Ethics committee of Federal state budgetary institution «V. A. Almazov National Medical Research Centre» of the Ministry of Health of the Russian Federation 197341. St. Petersburg, Ref-No. ABSTRACT No. 151; Federal State autonomous institution "National Scientific and Medical Center for Neurosurgery Named after the Academician N.N. Burdenko" of the Ministry of Health of the Russian Federation, Ref-No. 7710103758/771001001; Medical Ethics Committee Ziekenhuis Oost-Limburg, Ref-No. 15/091 U; B371201524198; Independent Ethis Committee IRCCS Instituto Clinico Humanitas, Ref-No. Prot. Nr. CE Humanitas ex D.M. 8/2/2013 161/17; Ethics Committee Di Area Vasta Emilia Centro Della Regione Emilia-Romagna, Ref-No. Ns. rif. 2019/PO/011 Vs. rif. Cod. CE: 17125; Romania Academy of Medical Sciences. The National Ethics Committee for Medicines and Medical Devices, Ref-No. 134 NP; University Hospitals Division, NHS Lothian, Queen’s Medical Research Institute, Ref-No. 2016/0231C, 16/NE/0244; ethics committee at the Lower Silesian Medical Chamber of Physicians in Wroclaw, Ref-No. 103/2018; ethics committee University of Regensburg, Ref-No. 16-171_2-103; ethics committee of the Chamber of Physicians of Westphalia-Lippe and the Westphalian Wilhelms University, Ref-No. 2016-236-b-S; Medical Association Bremen, ethics committee, Ref-No. RA / RE-533; State Chamber of Physicians of Thuringia, Ref-No. 59350/2017/114; ethic committee of Clínica La Sagrada Familia, Ref-No. Revision and Analysis Phenox - Protocol.
Acknowledgments
We would like to acknowledge the work of Lena Vadder, phenox GmbH.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @MartaAguilarPe2
Contributors ABo, MAP, HH, PL, CB, GG, SS, AS, LSt, FT, AP, CR, A-PN, XB, CL, ABe, LP, MM, TG, AC, JK, CWa, MDO, LSp, FC, SY, PK, NPN, SD, CWe acquired data for analysis. ABo and TW conceived the idea for the study and interpreted the data of work. ABo, PS, and TW drafted the manuscript, provided critical editing and guidance for the manuscript. ABo, PS, PL, LSt, CR, and TW approved the final version to be published. ABo and PS agreed to be accountable for all aspects of the work and ensured that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. ABo is the guarantor of this work.
Funding This study was funded by phenox GmbH, Bochum (Germany).
Competing interests ABo serves as a consultant for Balt, phenox, and Stryker. MAP serves as proctor and consultant for phenox. HH is a co-founder and shareholder of phenox, femtos, and CONTARA; serves as proctor and consultant for phenox; and declares personal financial interest in Johnson & Johnson and Penumbra. PL, CB, GG, SS, AS, AP, CR, A-PN, XB, AC, CWa, MDO, SY, NPN, CWe have no personal, financial, or institutional interest to report. LSt declares personal financial interest for Medtronic and Terumo. FT serves as a consultant for Balt and Medtronic. CL declares proctoring and consultant services for phenox and also serves as consultant for Penumbra and receives travel and meeting expanses from Acandis and Penumbra. ABe serves as consultant for Microvention and as CEC chairman for phenox for the ARTESp trial. LP declares consulting services for Balt, Cerus Endovascular, Microvention, Perflow, phenox, and Vesalio. MM serves as a proctor for Penumbra and declares support for Procardia and Polimed. TG serves as a proctor for phenox. JK serves as a proctor and consultant for Microvention and phenox with payment to his institution. LSp is a consultant for Balt, Medtronic, Microvention, phenox, and Stryker and also declares support for Balt, Medtronic, Microvention, and Stryker outside of this work. FC serves as a consultant for Balt, Medtronic, and Stryker; receives payment for support of Penumbra; and participates as DSMB/advisory board of Microvention. PK serves as proctor and consultant for phenox and received travel and meeting expenses from phenox. SD declares no other competing interests besides that he was the President of the Central and Eastern European Committee of ESMINT until September 2020. TW serves as consultant for phenox. PS serves as consultant for Penumbra, phenox, and Stryker.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.