Article Text

Abstract
Background: Cerebral infarction after aneurysmal subarachnoid haemorrhage (SAH) is presumed to be due to cerebral vasospasm, defined as arterial lumen narrowing from days 3 to 14.
Methods: We reviewed the computed tomography scans of 103 patients with aneurysmal SAH for radiographic cerebral infarction and controlled for other predictors of outcome. A blinded neuroradiologist reviewed the angiograms. Cerebral infarction from vasospasm was judged to be unlikely if it was visible on computed tomography within 2 calendar days of SAH or if angiography showed no vasospasm in a referable vessel, or both.
Results: Cerebral infarction occurred in 29 (28%) of 103 patients with SAH. 18 patients had cerebral infarction that was unlikely to be due to vasospasm because it was visible on computed tomography by day 2 (6 (33%)) or because angiography showed no vasospasm in a referable artery (7 (39%)), or both (5 (28%)). In a multivariate model, cerebral infarction was significantly related to World Federation of Neurologic Surgeons grade (odds ratio (OR) 1.5/grade, 95% confidence interval (CI) 1.1 to 2.01, p = 0.006) and SAH-Physiologic Derangement Score (PDS) >2 (OR 3.7, 95% CI 1.4 to 9.8, p = 0.01) on admission. Global cerebral oedema (OR 4.3, 95% CI 1.5 to 12.5, p = 0.007) predicted cerebral infarction. Patients with cerebral infarction detectable by day 2 had a higher SAH-PDS than patients with later cerebral infarction (p = 0.025).
Conclusions: : Many cerebral infarctions after SAH are unlikely to be caused by vasospasm because they occur too soon after SAH or because angiography shows no vasospasm in a referable artery, or both. Physiological derangement and cerebral oedema may be worthwhile targets for intervention to decrease the occurrence and clinical impact of cerebral infarction after SAH.
- MRI, magnetic resonance imaging
- PDS, Physiologic Derangement Score
- SAH, subarachnoid haemorrhage
- WFNS, World Federation of Neurologic Surgeons
Statistics from Altmetric.com
- MRI, magnetic resonance imaging
- PDS, Physiologic Derangement Score
- SAH, subarachnoid haemorrhage
- WFNS, World Federation of Neurologic Surgeons
Subarachnoid haemorrhage (SAH) is a common and serious disorder, affecting approximately 11 in every 100 000 people.1 Cerebral infarction after SAH is a well-described complication and is strongly associated with a poor outcome.2 Strategies to improve outcome after SAH usually focus on preventing or treating vasospasm.
Nimodipine reduces the risk of cerebral infarction after SAH due to vasospasm3; yet, cerebral infarction still occurs and remains a major predictor of poor outcome in multivariate models.4 Vasospasm is traditionally defined as arterial narrowing 3–14 days after SAH.5 Cerebral infarction after SAH is generally assumed to be due to vasospasm, but causality is difficult to prove in critically ill patients. Radiographic cerebral infarction allows different observers to independently review studies at leisure, but may not be symptomatic. Acute cerebral infarction may be due to the acute effects of haemorrhage and the spike in intracranial pressure, but there are few objectively defined risk factors. We sought to identify patients with cerebral infarction associated with SAH in whom the cerebral infarction was unlikely to be due to vasospasm and to describe novel risk factors for targeted intervention.
MATERIALS AND METHODS
Patients
We used a database of 103 aneurysmal patients with SAH admitted to our hospital from 2001 to 2004. The diagnosis of SAH was confirmed in the electronic medical record and with compatible computed tomography findings. To ensure complete data collection, all data were collected from an electronic medical record, and all patients had at least one computed tomograph archived on our electronic radiology system. Clinical variables collected included World Federation of Neurologic Surgeons (WFNS) grade on admission, age, sex, clinical history of hypertension (blood pressure >135/90 mm Hg or prescribed drugs for hypertension), coronary artery disease, tobacco use (>10 pack-years or current) and heart failure. The Northwestern University Institutional Review Board (Chicago, Illinois, USA) approved the project.
We corrected for admission physiologic derangement with the SAH-Physiologic Derangement Scale (PDS),6 a previously validated measure. The SAH-PDS is calculated as a score from 0 (least derangement) to 8 (most derangement) on the basis of first-measured alveolar–arterial oxygen tension gradient (+3 points for >125 mm Hg), sodium bicarbonate (+2 points for <20 mg/dl), serum glucose (+2 points for >180 mg/dl) and abnormal mean arterial pressure (+1 point for >130 or <70 mm Hg). All computed tomography scans of the head were electronically stored and were retrieved for review. A single trained reviewer rated the computed tomography scans for the SAH sum score,7 presence of thick clot (Fisher grade 3)8 and admission global cerebral oedema9 for each patient. The aneurysm site was classified as previously published.10
Neuro-intensive care unit management
Routine clinical management followed the most recent published clinical guidelines.11 All patients who spent ⩾48 h in an intensive care unit setting were managed with central venous pressure monitoring and repletion with crystalloid and colloid to a central venous pressure of at least 5 mm Hg. Nimodipine 60 mg was given every 4 h unless the patient was hypotensive. Hydrocephalus was treated with a ventricular drain. Hyperdynamic therapy, increasing volume status and using vasopressors to increase blood pressure to maximise cerebral perfusion and cardiac preload, with or without angiographic intervention, was used for symptomatic vasospasm. Except for the patients with classic perimesencephalic haemorrhage,12 all patients underwent angiography as soon as they were haemodynamically stable, and coiling or clipping of the offending aneurysm if feasible. We defined “acute”, “intermediate” and “late” repair as previously published.13 Surgical procedures were performed by fellowship-trained vascular neurosurgeons, and endovascular procedures were performed by fellowship-trained neuroradiologists or endovascular neurosurgeons. Angiograms were repeated for new focal neurological signs suggestive of vasospasm, or for new computed tomography findings suggestive of ischaemia.
Acquisition of computed tomography scans
Computed tomography scans were acquired on admission, after placement of a ventricular catheter or intracranial pressure monitor, and after endovascular or neurosurgical intervention. Computed tomography scans were repeated for any change in neurological status, and after the ventricular drain was removed.
Definition of cerebral infarction
The diagnosis of cerebral infarction was made on review of all available computed tomography scans (up to 14 days after haemorrhage) by a single board-certified vascular neurologist with certificate of added qualifications-certified neuroradiologist confirmation. We did not include hypodensities that cleared by 14 days after SAH (probably representing focal oedema from surgical retraction), lesions that were due to ventricular drain mishap, or resolving intracerebral haemorrhage. We counted the day of occurrence as the first day a focal hypodensity could clearly be seen on the computed tomography scan of the head, from day 0 (the day of SAH) until day 14. Cerebral infarction was not defined by symptoms.
Every patient with a cerebral infarction had a review of all angiograms by a certificate of added qualifications-certified neuroradiologist blinded to clinical information and computed tomography scan results. All angiograms were performed in a modern digital angiogram suite equipped with full electronic storage and retrieval. We analysed angiograms rather than transcranial Doppler values because angiography is considered to be the reference standard for vasospasm. The clinical database and computed tomography readings were locked before angiogram review to avoid bias. Vasospasm was scored as mild (<40%), moderate (40–60%) or severe (>60%) narrowing by comparison to a normal segment and to the baseline angiogram. Location of cerebral infarction was referenced to the anterior cerebral artery middle cerebral artery, or posterior cerebral artery. Cerebral infarction was recorded as left, right or bilateral. We recorded agreement between cerebral infarction and angiographic vasospasm, and between cerebral infarction and location of the ruptured aneurysm. We considered cerebral infarction from vasospasm unlikely if it was detectable on computed tomography by day 2 (within 2 calendar days of aneurysm rupture) or if no angiographic vasospasm was seen in the arterial distribution of cerebral infarction.
Statistical methods
Data were collected on a standardised form and entered into a custom database. All data were double checked at the time of collection, and cleaned and logically checked at the time of data entry. Statistical analyses were performed with commercially available software (SPSS V.13). Comparisons between two non-normally distributed variables were performed with a Mann–Whitney U or Kruskal–Wallis H test as appropriate, and normally distributed variables were compared with analysis of variance. Logistic regression was used to predict cerebral infarction. Variables were added one at a time with a forward-conditional method (p<0.1 to enter). Non-normal variables (WFNS, SAH sum score per quartile) were treated as ordinal variables. We considered p⩽0.05 significant.
RESULTS
Table 1 shows the demographic characteristics of the patients. Cerebral infarction occurred in 29 of 103 (28%) patients. There were five incident cerebral infarctions visible on computed tomography on day 0 (the day of haemorrhage), one on day 1, five on day 2, four on days 3 and 4, and ten after day 4 (fig 1). Sixteen patients died in hospital.
Demographic characteristics

Life table plot of time to stroke or death (censored) of the 36 patients with stroke.
Eight patients were admitted to our institution at least two days after SAH, and 27 patients were admitted the day after SAH. Of the patients who were transferred to us more than 1 day after SAH, only one had a cerebral infarction, which occurred 3 days after admission.
Of the 29 patients with cerebral infarction, 25 patients underwent 41 angiograms an average of 3.7 days after SAH; in 15 patients two angiograms were performed. Four WFNS grade 5 patients had no catheter angiogram because of haemodynamic instability or neurological catastrophe. In 20 of the 41 angiograms no angiographic vasospasm was identified. Nine patients with cerebral infarction did not have vasospasm identified on any angiogram. There was 72% agreement between angiographic vasospasm and radiographic cerebral infarction when the tests were performed within 2 calendar days, and ⩽60% when they were separated by 3 or more days (p = 0.2). Any radiographic cerebral infarction was in the same arterial distribution as the aneurysm in only 14 (48%) patients; in 8 (28%) patients cerebral infarction was bilateral or contralateral to the aneurysm.
Four patients had cerebral infarction 3–7 days before aneurysm obliteration, three on the same day as aneurysm obliteration, four patients on 1, 2 or 3 days after aneurysm obliteration, and the remainder on 4 or more days after aneurysm obliteration.
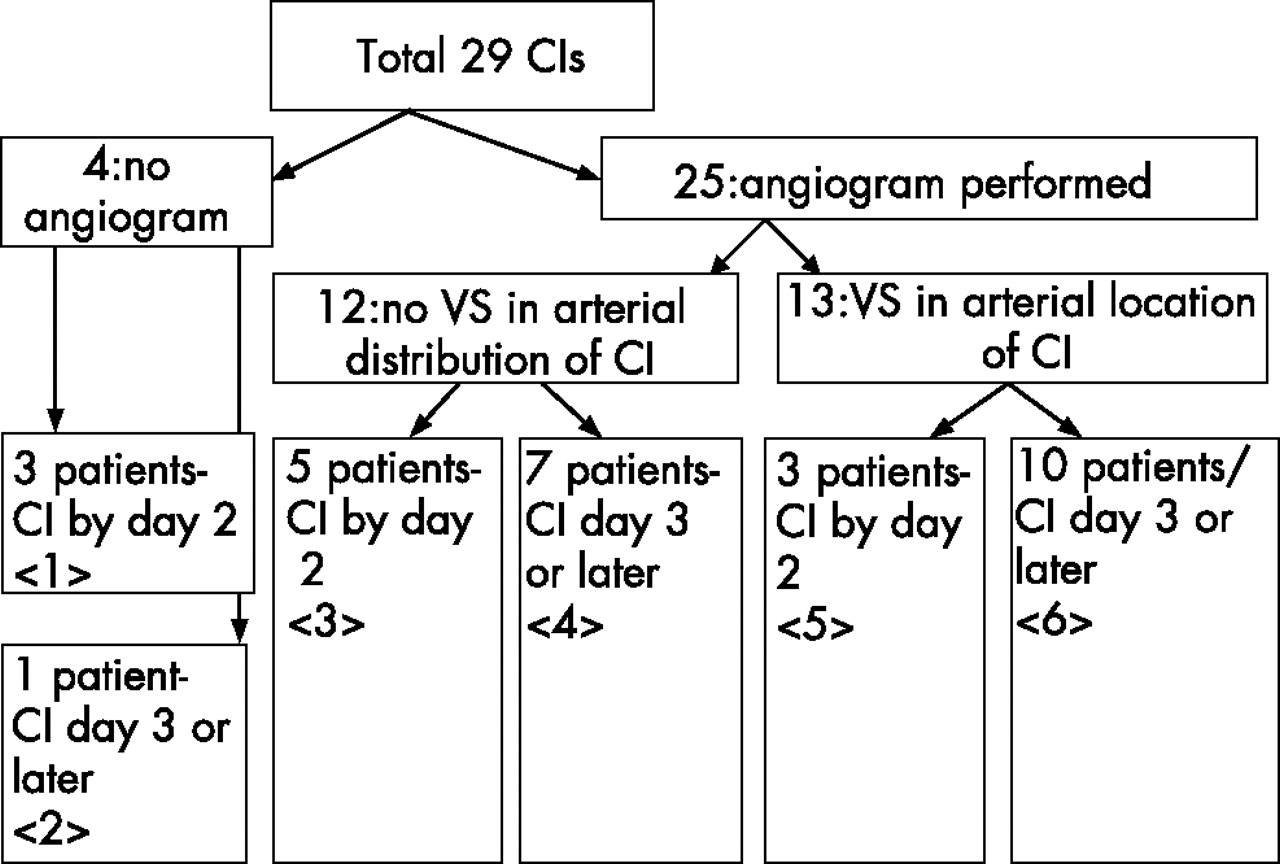
Figure 2 shows the breakdown of patients with cerebral infarction by angiogram and timing of cerebral infarction. In all, 18 patients had cerebral infarction that was unlikely to be due to vasospasm, because it was visible on computed tomography by day 2 (boxes <1> and <5>, 6 (33%) patients)or because angiography showed no vasospasm in a referable artery (box <4>, 7 (38%) patients), or both (box <3>, 5(27%) patients). Patients with cerebral infarction visible on computed tomography by day 2 had more abnormal SAH-PDS scores at admission (p = 0.025) than patients whose cerebral infarction was detected on day 3 and later (table 2), especially scores >2.
Characteristics of all cerebral infarctions (CI), CI without angiographic vasospasm in a referable vessel and CI visible on computed tomography on day 2
{kind=link}
{kind=link}
Diagram depicting the breakdown of the 29 patients with cerebral infarction (CI). Eighteen patients had CI that was unlikely to be due to vasospasm because it was visible on computed tomography by day 2 (6 (33%)) or because angiography showed no vasospasm in a referable artery (7 (38%)), or both (5 (27%)). Four patients with World Federation of Neurologic Surgeons grade 5 did not undergo angiography because of haemodynamic instability or neurological catastrophe, three of whom had cerebral infarction detectable by day 2. CI, cerebral infarction, VS, vasospasm. Boxes are marked with <number> for reference to the text.
Table 3 shows the univariate predictors of cerebral infarction. In logistic regression, SAH-PDS ⩾2 (odds ratio (OR) = 3.7, 95% confidence interval (CI) 1.4 to 9.8, p = 0.01) and WFNS grade (OR = 1.5/grade, 95% CI 1.1 to 2.01, p = 0.006) were associated with radiographic cerebral infarction. WFNS and SAH-PDS were correlated (p = 0.001), but there was no interaction in the model. When the 11 patients with cerebral infarction visible by day 2 were excluded, WFNS (OR 1.5/grade, 95% CI 1.05 to 2.2, p = 0.027) and global cerebral oedema on admission (OR 3.8, 95% CI 1.02 to 14.1, p = 0.046) were found to be associated with radiographic cerebral infarction in logistic regression. Global cerebral oedema was not associated with angiographic vasospasm (p>0.2).
Variables associated with cerebral infarction
Aneurysm location on angiography was associated with cerebral infarction (p = 0.003; table 4), with an increased risk of cerebral infarction with posterior circulation and intracranial aneurysms, as compared with middle and anterior cerebral artery aneurysms.
Site of aneurysm related to cerebral infarction
Patients without an aneurysm on angiography had a low risk of cerebral infarction. Controlling for posterior aneurysm location or no identifiable aneurysm did not add to the logistic regression models presented above.
Of the 80 patients in whom aneurysm obliteration was feasible, 69 had urgent repair, nine had intermediate repair and only two had late repair. In all, 62 (78%) patients had surgical clipping and 18 (22%) had endovascular coiling. There were no differences in neurological grade, age, SAH-PDS, global cerebral oedema or mortality with cerebral infarction before aneurysm obliteration, or bilateral cerebral infarction.
DISCUSSION
These data suggest that vasospasm, as traditionally defined, may contribute less to cerebral infarction after SAH than was previously thought. More than one third of cerebral infarctions were visible on computed tomography by day 2, making vasospasm an unlikely cause. More than one quarter of cerebral infarctions were in a vascular distribution unaffected by angiographic vasospasm. We found that patients with cerebral infarction detectable on computed tomography by day 2 had more abnormal SAH-PDS scores, especially scores >2. SAH-PDS >2 was no longer significant in multivariate models after cerebral infarction visible by day 2 was excluded, underscoring the importance of physiological derangement in early cerebral infarction. More abnormal acute physiology may indicate a greater risk of cerebral infarction during aneurysm obliteration and greater physiological derangement from cardiovascular effects of haemorrhage. Alternative mechanisms for cerebral infarction include arterial embolism, hypotension and focal neuronal toxicity due to metabolic derangements (ie, hyperglycaemia, acidaemia and hypoxia).
Two thirds of patients with SAH have multiple causes for cerebral infarction, and distinguishing them from one another may be impossible. It is possible that modern therapy for vasospasm—nimodipine prophylaxis, transcranial Doppler screening, treatment with hyperdynamic therapy and angioplasty—has had a major effect and the residual cerebral infarctions are due to other causes. The use of statins before14 and during15 hospitalisation is likely to further reduce the risks of cerebral infarction and vasospasm.
We looked for cerebral infarctions that were unlikely to be due to vasospasm by virtue of timing, angiography, or both. We propose that cerebral infarction detectable on computed tomography by day 2 is unlikely to be caused by vasospasm but may be particularly related to admission physiological derangement and neurological grade. Cerebral infarction detectable from days 3 to 14 without angiographic vessel narrowing may not be related to vasospasm, and cerebral infarction detectable from days 3 to 14 in the presence of mildly narrowed arterial supply is probably related to vasospasm. The lack of a gold standard for cerebral infarction due to vasospasm makes aetiology difficult to attribute with total confidence.
Our data show that acute physiological derangement is a powerful predictor of later cerebral infarction. Derived from the Acute Physiology And Chronic Health Evaluation (APACHE) score,16 each factor in the SAH-PDS quantifies a specific cause of neurotoxicity: acidaemia, impaired oxygen delivery, hyperglycaemia and abnormal brain perfusion. Aggressive correction of these abnormalities may be a promising future approach for the prevention of cerebral infarction. The role of later SAH-PDS scores and physiological derangement on cerebral infarction and outcome after SAH needs further study.
The presence of global cerebral oedema on admission probably represents a sudden loss of cerebral blood flow at the time of haemorrhage, with neuronal swelling, so its strong association with subsequent cerebral infarction is reasonable. Its association with poor outcome, but not with cerebral infarction, has been previously reported.9 We found it to be associated with cerebral infarction after 2 days following statistical correction for WFNS.
High blood pressure after SAH is related to ultra-early rebleeding,17 and acute blood pressure reduction to prevent rebleeding before aneurysm repair may decrease cerebral perfusion and cause cerebral infarction. Unfortunately, we did not have access to continuous blood pressure data to test this hypothesis. A history of hypertension may no longer predict cerebral infarction because of increased sensitivity to the risks of rapidly reducing blood pressure. Increasing neurological grade,18 the most important variable in our data, relates to neurological grade as a marker of haemorrhage severity. Good-grade patients have a low risk of cerebral infarction after early surgery.19
Acute surgery has been associated with an increased frequency of cerebral infarction,13,20 perhaps because the brain is more sensitive to surgical and endovascular manipulation immediately after SAH. Nearly all patients in our series had acute aneurysm obliteration. Early repair is still beneficial because the higher risk of cerebral infarction is offset by a lower risk of aneurysm rebleeding.21,22
We were not able to confirm that thick clot (Fisher grade 3)23 or intraventricular haemorrhage increased the risk of cerebral infarction,24 although the amount of subarachnoid blood25 and worse neurological grade did increase the risk.18 This may reflect a benefit from the ability to wash out subarachnoid blood at surgery,26 the predominant mode of treatment at our institution. Thick clot and ventricular haemorrhage may predict cerebral infarction during the vasospasm period but not early cerebral infarction. Nimodipine reduces the risk of cerebral infarction from vasospasm from increased subarachnoid blood,3 and some previous series were completed before widespread nimodipine use, or when fluid restriction and antifibrinolytics25 were still in use, both of which increase the risk for cerebral infarction from vasospasm. We were not able to test for some other previously reported associations, such as nocturnal aneurysm rupture, loss of consciousness or duration of arterial occlusion during surgery.27 Our sample size may have been too small to confirm some previously reported observations.
Our sample contained a relatively high proportion of patients in a coma and with posterior circulation aneurysms. This probably represents a referral bias, and our results may not apply to a community sample of patients with SAH. Specific aneurysm locations were not associated with cerebral infarction when individually tested, although, overall, aneurysm location and cerebral infarction were associated. We may have had reduced power to show these expected associations in subgroup analyses, or this may reflect our institutional expertise in treating posterior circulation aneurysms.
Cerebral infarction did not increase the risk of death, but a small to moderate cerebral infarction might leave a patient alive but disabled. Treatments that prevent cerebral infarction but not death2 have not gained wide acceptance.
Several limitations to our data deserve mention. We were unable to correlate cerebral infarction with a standardised neurological disability scale, although there is often concordance between the computed tomography and clinical picture. Future research should compare serial standardised neurological examinations (eg, National Institutes of Health Stroke Scale) with radiographic cerebral infarction. These patients were retrospectively identified, and the relationships we describe should be prospectively confirmed. These data do not allow us to determine the exact mechanism for cerebral infarction in each case, which is often multifactorial. We used computed tomography to define cerebral infarction, although magnetic resonance imaging (MRI) is much more sensitive.28,29 MRI would probably show more cerebral infarctions, although acute MRIs are difficult to obtain in critically ill patients. The fogging effect may have led to some false-negative follow-up computed tomography scans, and MRI or later computed tomography might be necessary to detect this.
In summary, vasospasm, as traditionally defined, does not explain the significant number of cerebral infarctions after SAH that occur within 2 days of haemorrhage, or are not associated with angiographic vasospasm, or both. Acute physiological derangement (SAH-PDS>2) is related to cerebral infarction that is detectable on computed tomography by day 2. Athough we must continue to prevent and aggressively treat vasospasm, further reductions in morbidity after SAH are likely to require complementary approaches. Targeted, aggressive intensive care management of acute physiological derangement and global cerebral oedema on admission is a promising approach for reducing the incidence of cerebral infarction after SAH.
REFERENCES
Footnotes
-
Published Online First 4 July 2006
-
This research was funded departmentally.
-
Competing interests: None.
-
The Northwestern University Institutional Review Board approved the project.
Linked Articles
- Editorial commentary